Department of Animal Molecular Biology, National Research Institute of Animal Production, Krakowska 1 St, Balice, 32-083, Poland.
Center for Experimental and Innovative Medicine, University of Agriculture in Krakow, Redzina 1c, Krakow, 30-248, Poland.
Mamm Genome. 2024 Dec;35(4):600-620. doi: 10.1007/s00335-024-10057-0. Epub 2024 Aug 14.
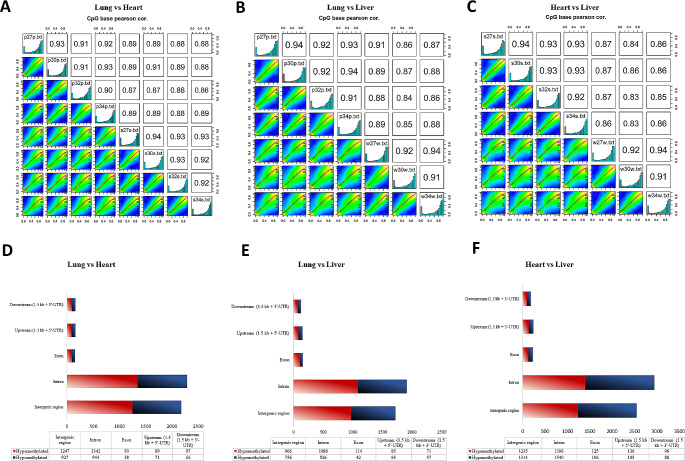
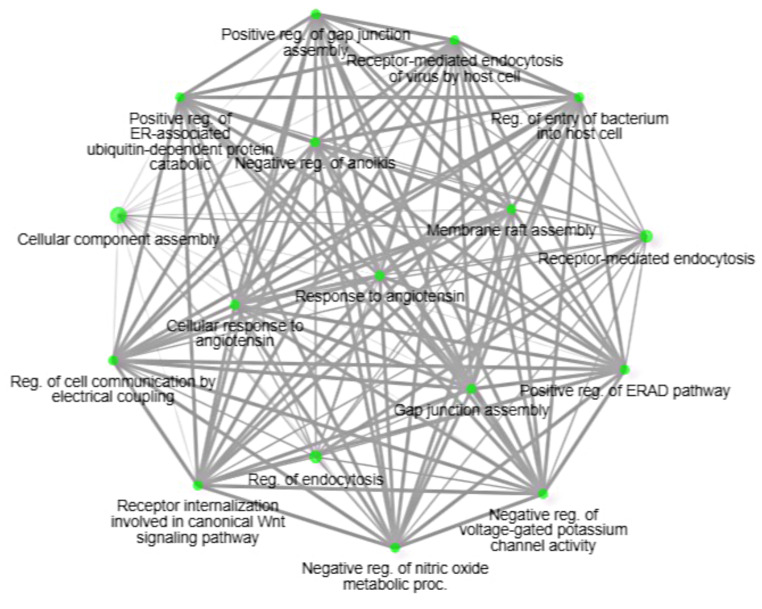
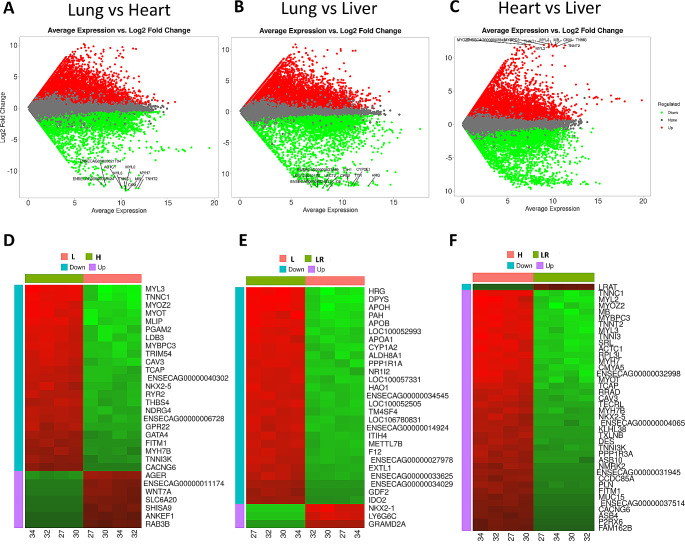
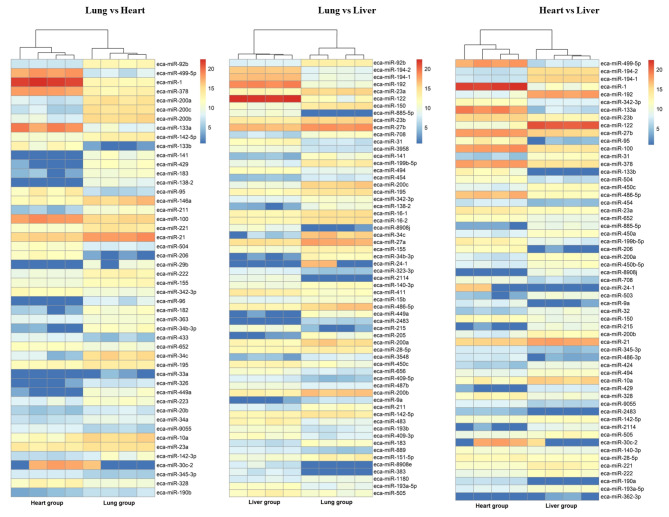
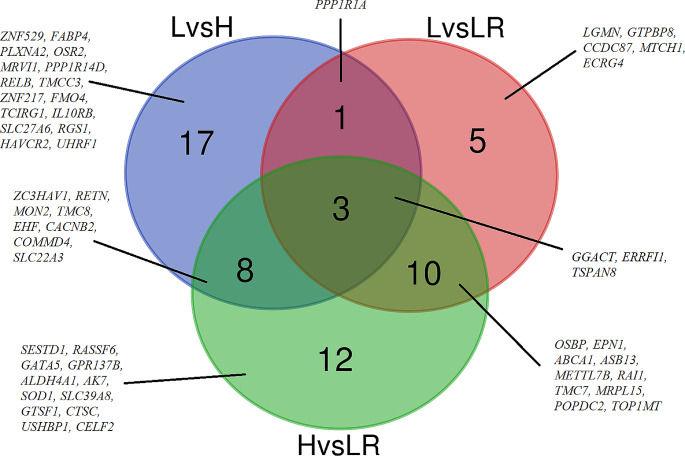
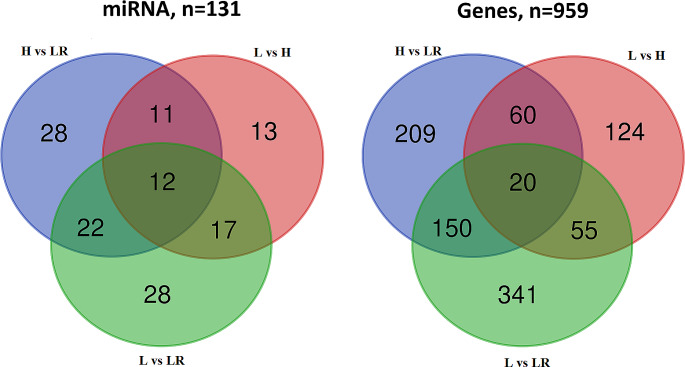
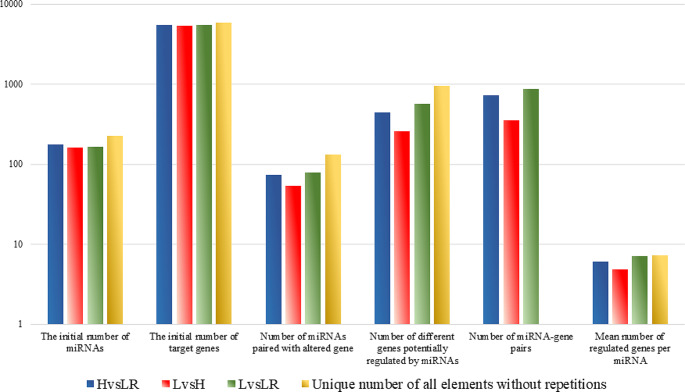
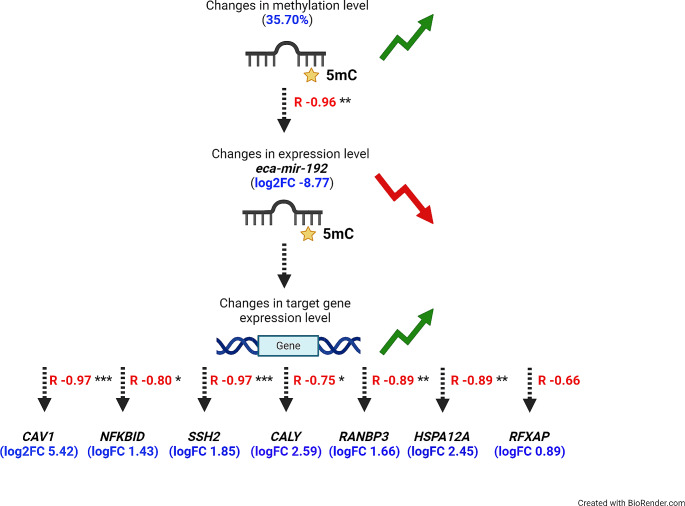
DNA methylation and microRNA (miRNA) expression are epigenetic mechanisms essential for regulating tissue-specific gene expression and metabolic processes. However, high-resolution transcriptome, methylome, or miRNAome data is only available for a few model organisms and selected tissues. Up to date, only a few studies have reported on gene expression, DNA methylation, or miRNA expression in adult equine tissues at the genome-wide level. In the present study, we used RNA-Seq, miRNA-seq, and reduced representation bisulfite sequencing (RRBS) data from the heart, lung, and liver tissues of healthy cold-blooded horses to identify differentially expressed genes (DEGs), differentially expressed miRNA (DE miRNA) and differentially methylated sites (DMSs) between three types of horse tissues. Additionally, based on integrative omics analysis, we described the observed interactions of epigenetic mechanisms with tissue-specific gene expression alterations. The obtained data allowed identification from 4067 to 6143 DMSs, 9733 to 11,263 mRNAs, and 155 to 185 microRNAs, differentially expressed between various tissues. We pointed out specific genes whose expression level displayed a negative correlation with the level of CpG methylation and miRNA expression and revealed biological processes that they enrich. Furthermore, we confirmed and validated the accuracy of the Next-Generation Sequencing (NGS) results with bisulfite sequencing PCR (BSP) and quantitative PCR (qPCR). This comprehensive analysis forms a strong foundation for exploring the epigenetic mechanisms involved in tissue differentiation, especially the growth and development of the equine heart, lungs, and liver.
DNA 甲基化和 microRNA(miRNA)表达是调控组织特异性基因表达和代谢过程的重要表观遗传机制。然而,高分辨率转录组、甲基组或 miRNA 组数据仅可用于少数模式生物和选定组织。迄今为止,只有少数研究报道了成年冷血马组织在全基因组水平上的基因表达、DNA 甲基化或 miRNA 表达。在本研究中,我们使用了来自健康冷血马心脏、肺和肝脏组织的 RNA-Seq、miRNA-seq 和简化代表性亚硫酸氢盐测序(RRBS)数据,以鉴定三种马组织之间差异表达的基因(DEGs)、差异表达的 miRNA(DE miRNA)和差异甲基化位点(DMSs)。此外,基于综合组学分析,我们描述了观察到的表观遗传机制与组织特异性基因表达改变之间的相互作用。获得的数据允许从 4067 到 6143 个 DMS、9733 到 11263 个 mRNA 和 155 到 185 个 miRNA 之间鉴定出差异表达,这些差异表达存在于不同的组织中。我们指出了一些表达水平与 CpG 甲基化和 miRNA 表达呈负相关的特定基因,并揭示了它们富集的生物学过程。此外,我们通过亚硫酸氢盐测序 PCR(BSP)和定量 PCR(qPCR)对下一代测序(NGS)结果进行了验证和验证。这项综合分析为探索组织分化中涉及的表观遗传机制,特别是马心脏、肺和肝脏的生长和发育提供了坚实的基础。