Szabó P Bernát, Schätzle Zeno, Entwistle Michael T, Noé Frank
Department of Mathematics and Computer Science, FU Berlin, Arnimallee 6, Berlin 14195, Germany.
Microsoft Research AI4Science, Karl-Liebknecht Str. 32, Berlin 10178, Germany.
J Chem Theory Comput. 2024 Aug 30;20(18):7922-35. doi: 10.1021/acs.jctc.4c00678.
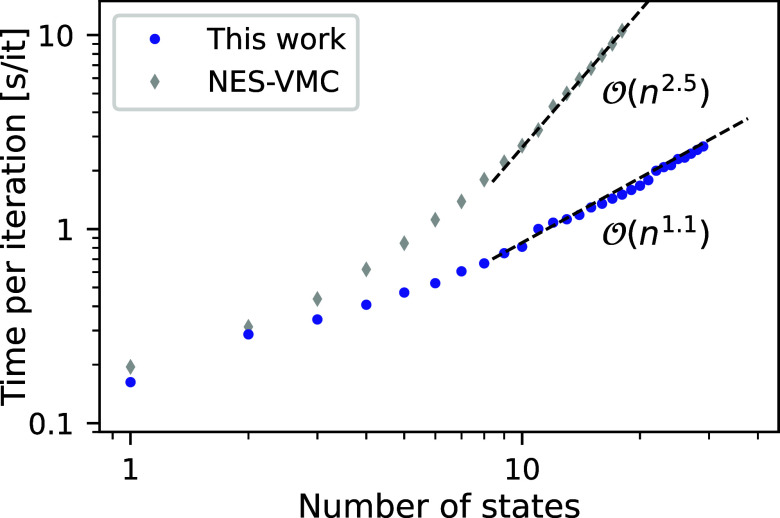
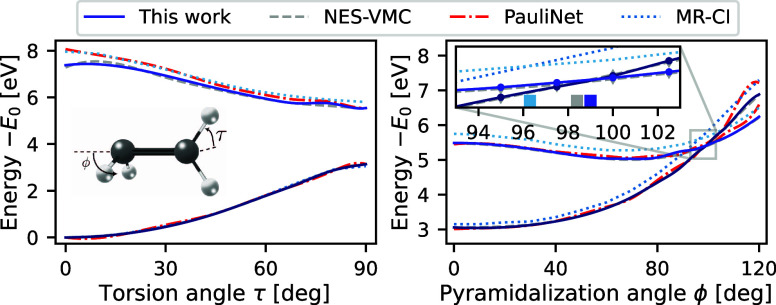
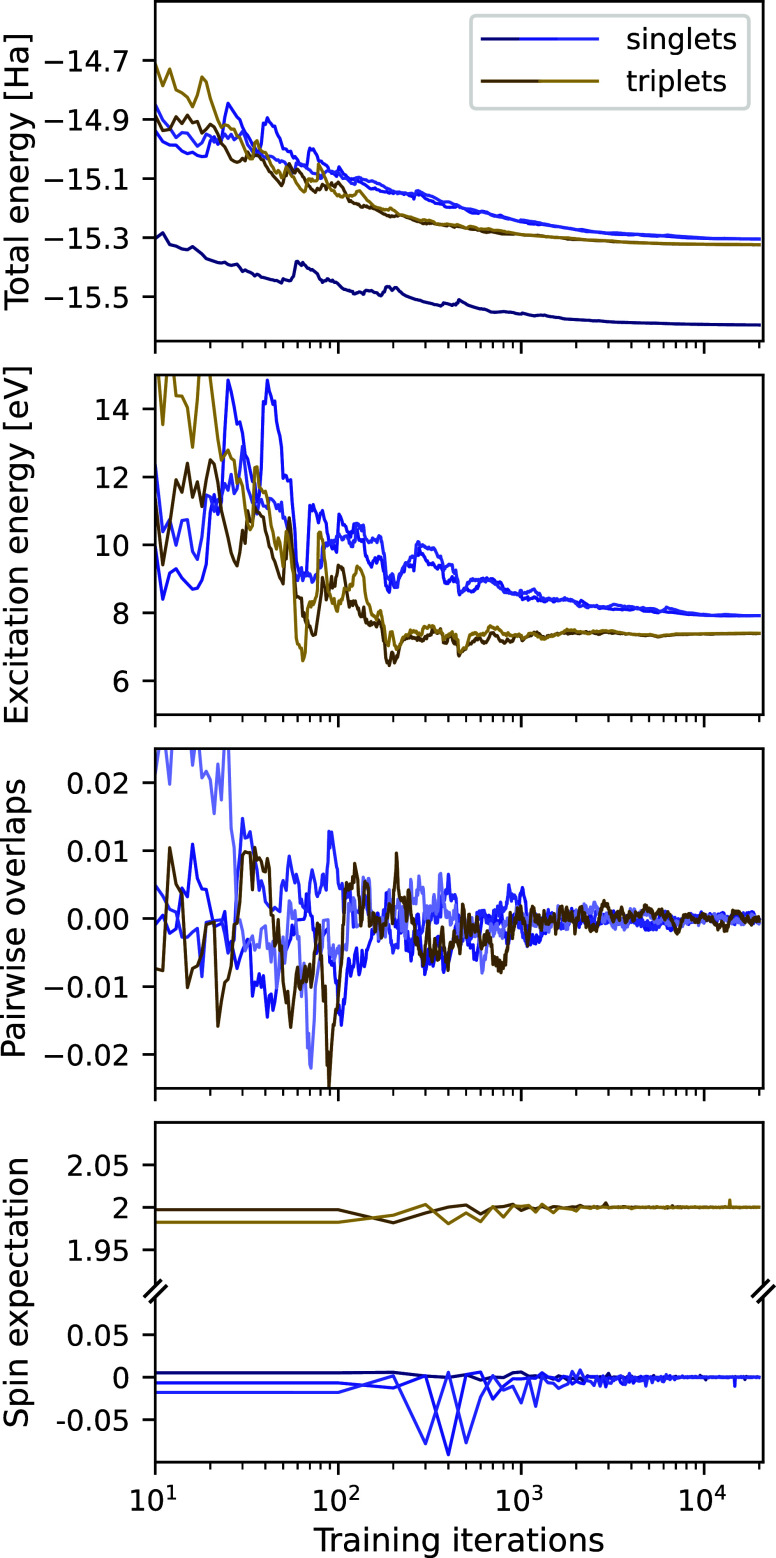
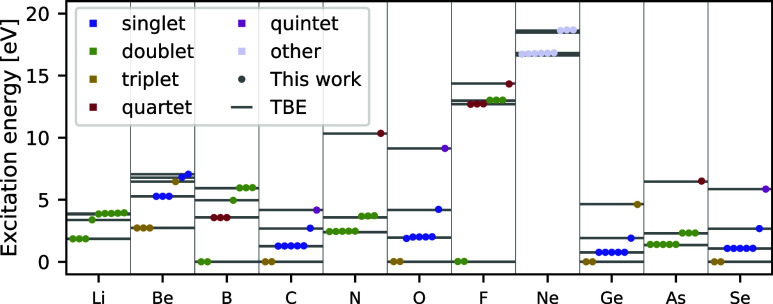
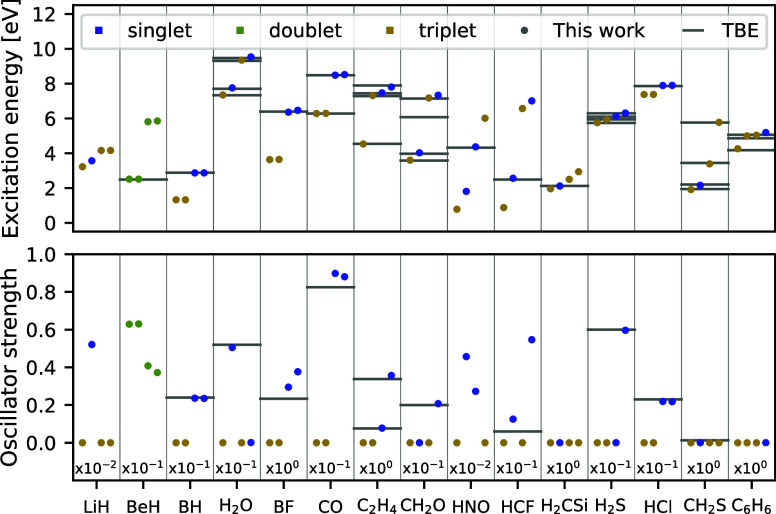
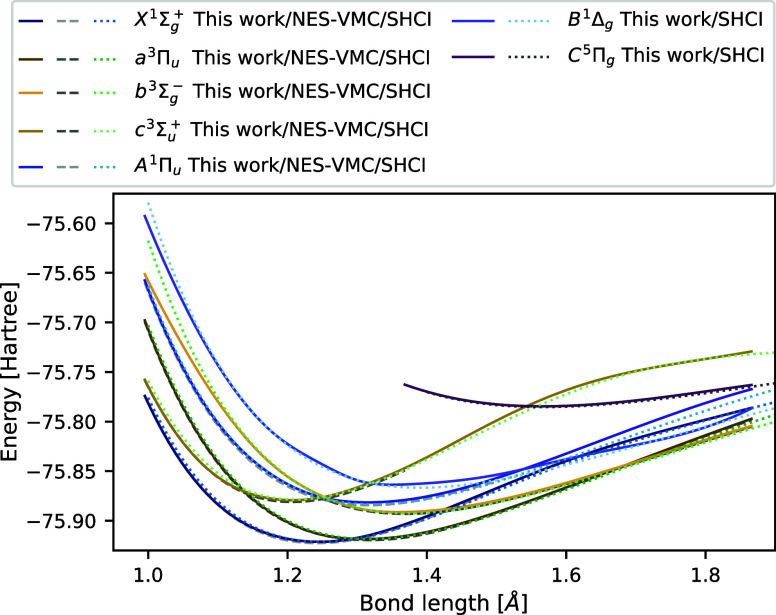
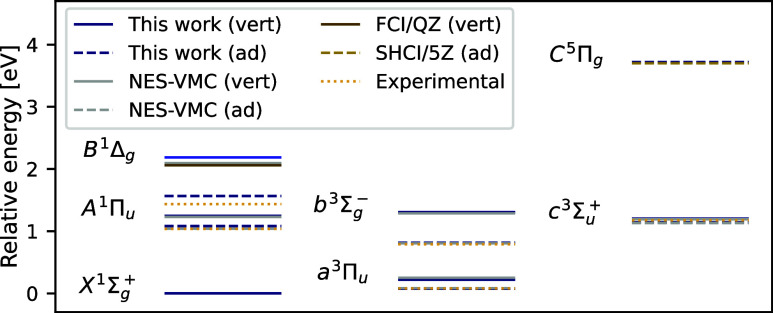
We introduce several improvements to the penalty-based variational quantum Monte Carlo (VMC) algorithm for computing electronic excited states of Entwistle et al. [Nat. Commun. , 274 (2023)] and demonstrate that the accuracy of the updated method is competitive with other available excited-state VMC approaches. A theoretical comparison of the computational aspects of these algorithms is presented, where several benefits of the penalty-based method are identified. Our main contributions include an automatic mechanism for tuning the scale of the penalty terms, an updated form of the overlap penalty with proven convergence properties, and a new term that penalizes the spin of the wave function, enabling the selective computation of states with a given spin. With these improvements, along with the use of the latest self-attention-based ansatz, the penalty-based method achieves a mean absolute error below 1 kcal/mol for the vertical excitation energies of a set of 26 atoms and molecules, without relying on variance matching schemes. Considering excited states along the dissociation of the carbon dimer, the accuracy of the penalty-based method is on par with that of natural-excited-state (NES) VMC, while also providing results for additional sections of the potential energy surface, which were inaccessible with the NES method. Additionally, the accuracy of the penalty-based method is improved for a conical intersection of ethylene, with the predicted angle of the intersection agreeing well with both NES-VMC and multireference configuration interaction.
我们对基于惩罚的变分量子蒙特卡罗(VMC)算法进行了多项改进,用于计算Entwistle等人[《自然·通讯》,274(2023)]的电子激发态,并证明更新方法的准确性与其他可用的激发态VMC方法具有竞争力。本文对这些算法的计算方面进行了理论比较,确定了基于惩罚方法的几个优点。我们的主要贡献包括一种自动调整惩罚项尺度的机制、一种具有已证明收敛性质的重叠惩罚的更新形式,以及一个惩罚波函数自旋的新项,从而能够选择性地计算具有给定自旋的状态。通过这些改进,以及使用最新的基于自注意力的近似,基于惩罚的方法在不依赖方差匹配方案的情况下,对于一组26个原子和分子的垂直激发能实现了低于1 kcal/mol的平均绝对误差。考虑到碳二聚体解离过程中的激发态,基于惩罚的方法的准确性与自然激发态(NES)VMC相当,同时还提供了势能面其他部分的结果,而这些结果是NES方法无法获得的。此外,基于惩罚的方法对于乙烯的锥形交叉点的准确性有所提高,预测的交叉点角度与NES-VMC和多参考组态相互作用都吻合得很好。