Department of Women's and Children's Health, Childhood Cancer Research Unit, Karolinska Institutet, Stockholm, 171 77, Sweden.
Department of Immunology, Genetics, and Pathology, Uppsala University, Uppsala, 752 36, Sweden.
Mol Cancer. 2024 Aug 31;23(1):180. doi: 10.1186/s12943-024-02091-y.
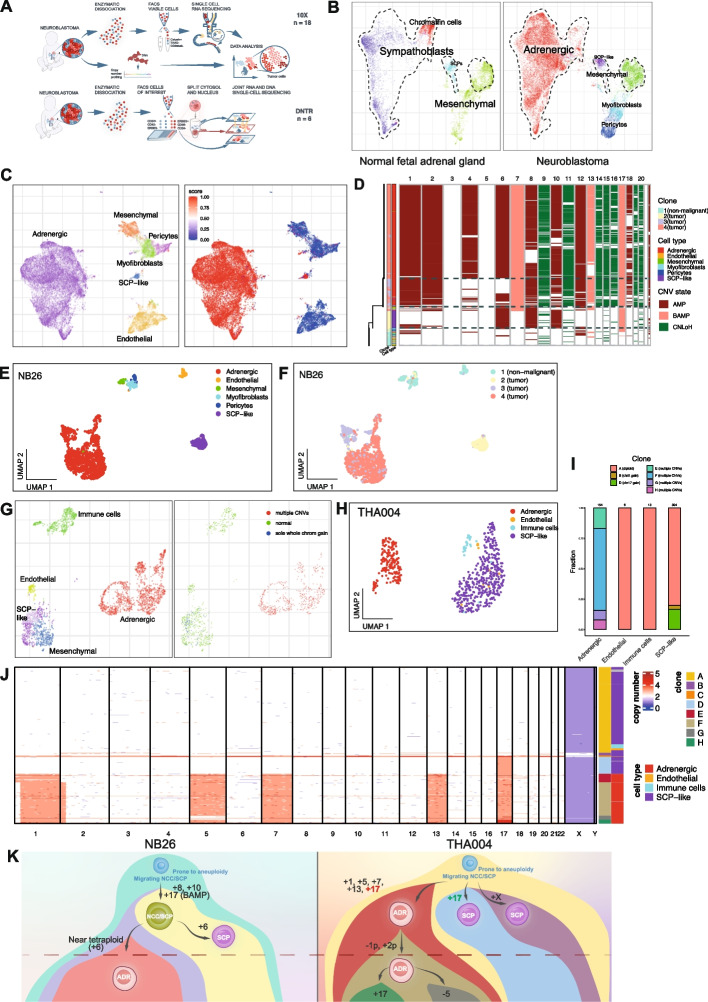
Neuroblastoma (NB) is a heterogeneous embryonal malignancy and the deadliest tumor of infancy. It is a complex disease that can result in diverse clinical outcomes. In some children, tumors regress spontaneously. Others respond well to existing treatments. But for the high-risk group, which constitutes approximately 40% of all patients, the prognosis remains dire despite collaborative efforts in basic and clinical research. While its exact cellular origin is still under debate, NB is assumed to arise from the neural crest cell lineage including multipotent Schwann cell precursors (SCPs), which differentiate into sympatho-adrenal cell states eventually producing chromaffin cells and sympathoblasts.
To investigate clonal development of neuroblastoma cell states, we performed haplotype-specific analysis of human tumor samples using single-cell multi-omics, including joint DNA/RNA sequencing of sorted single cells (DNTR-seq). Samples were also assessed using immunofluorescence stainings and fluorescence in-situ hybridization (FISH).
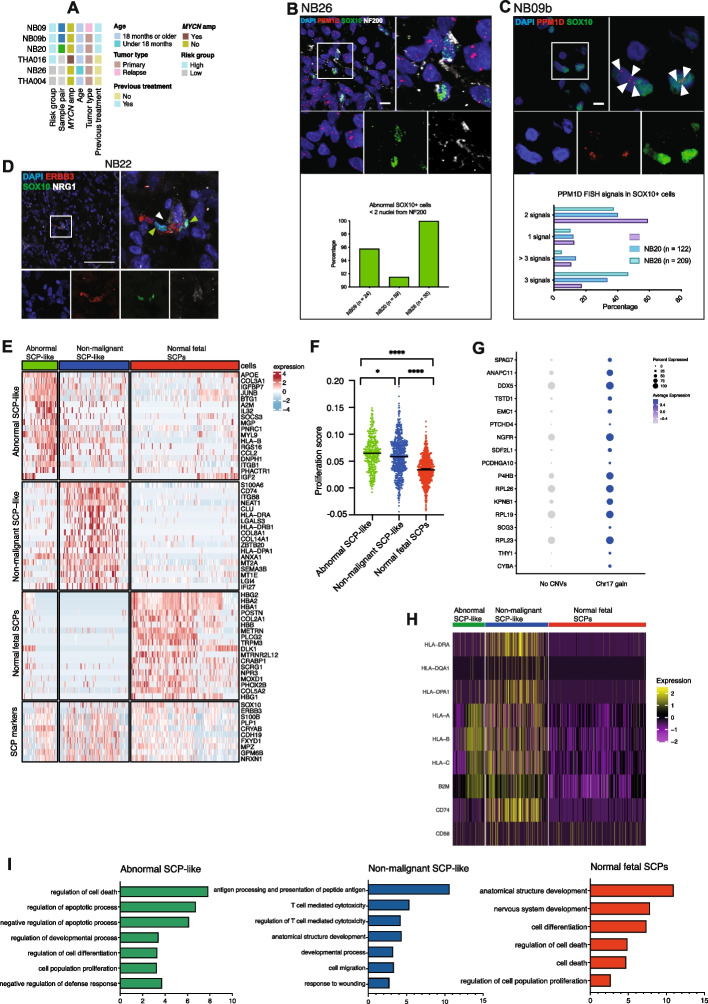
Beyond adrenergic tumor cells, we identify subpopulations of aneuploid SCP-like cells, characterized by clonal expansion, whole-chromosome 17 gains, as well as expression programs of proliferation, apoptosis, and a non-immunomodulatory phenotype.
Aneuploid pre-malignant SCP-like cells represent a novel feature of NB. Genetic evidence and tumor phylogeny suggest that these clones and malignant adrenergic populations originate from aneuploidy-prone cells of migrating neural crest or SCP origin, before lineage commitment to sympatho-adrenal cell states. Our findings expand the phenotypic spectrum of NB cell states. Considering the multipotency of SCPs in development, we suggest that the transformation of fetal SCPs may represent one possible mechanism of tumor initiation in NB with chromosome 17 aberrations as a characteristic element.
神经母细胞瘤(NB)是一种异质性胚胎恶性肿瘤,也是婴儿期最致命的肿瘤。它是一种复杂的疾病,可导致不同的临床结果。在一些儿童中,肿瘤会自发消退。其他儿童对现有治疗方法反应良好。但是,对于约占所有患者 40%的高危组,尽管在基础和临床研究方面进行了协作努力,但预后仍然很差。虽然其确切的细胞起源仍存在争议,但 NB 被认为起源于神经嵴细胞谱系,包括多能 Schwann 细胞前体(SCP),这些细胞最终分化为交感肾上腺细胞状态,产生嗜铬细胞和交感母细胞。
为了研究神经母细胞瘤细胞状态的克隆发育,我们使用单细胞多组学(包括对分选的单细胞进行联合 DNA/RNA 测序(DNTR-seq))对人肿瘤样本进行单倍型特异性分析。还使用免疫荧光染色和荧光原位杂交(FISH)评估了样本。
除了肾上腺素能肿瘤细胞外,我们还鉴定了具有克隆性扩张、整条 17 号染色体增益以及增殖、凋亡和非免疫调节表型表达程序的非整倍体 SCP 样细胞亚群。
非整倍体恶性前 SCP 样细胞是 NB 的一个新特征。遗传证据和肿瘤系统发育表明,这些克隆和恶性肾上腺素能群体起源于迁移的神经嵴或 SCP 起源的非整倍体易位细胞,然后再向交感肾上腺细胞状态分化。我们的发现扩展了 NB 细胞状态的表型谱。考虑到 SCP 在发育中的多能性,我们认为胎儿 SCP 的转化可能是 NB 中以染色体 17 异常为特征的肿瘤起始的一种可能机制。