AG Algorithmic Bioinformatics, Leibniz-Institut für Immuntherapie, Regensburg 93053, Germany.
deCODE genetics/Amgen Inc, Reykjavik 101, Iceland.
Bioinformatics. 2024 Sep 1;40(Suppl 2):ii11-ii19. doi: 10.1093/bioinformatics/btae391.
Complex structural variants (SVs) are genomic rearrangements that involve multiple segments of DNA. They contribute to human diversity and have been shown to cause Mendelian disease. Nevertheless, our abilities to analyse complex SVs are very limited. As opposed to deletions and other canonical types of SVs, there are no established tools that have explicitly been designed for analysing complex SVs.
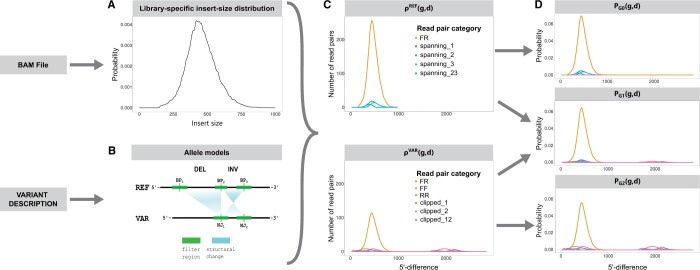
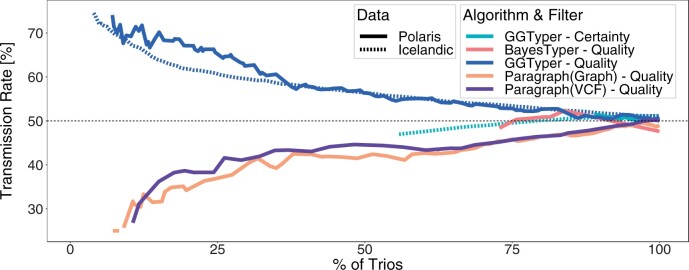
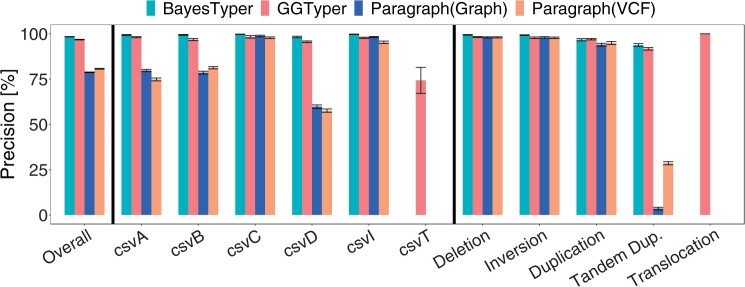
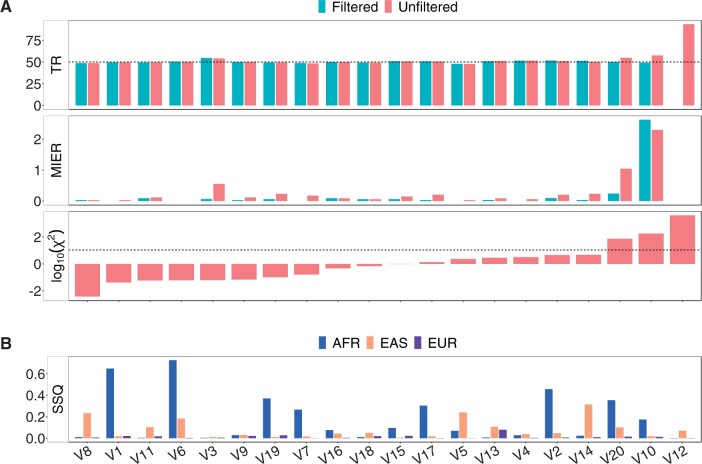
Here, we describe a new computational approach that we specifically designed for genotyping complex SVs in short-read sequenced genomes. Given a variant description, our approach computes genotype-specific probability distributions for observing aligned read pairs with a wide range of properties. Subsequently, these distributions can be used to efficiently determine the most likely genotype for any set of aligned read pairs observed in a sequenced genome. In addition, we use these distributions to compute a genotyping difficulty for a given variant, which predicts the amount of data needed to achieve a reliable call. Careful evaluation confirms that our approach outperforms other genotypers by making reliable genotype predictions across both simulated and real data. On up to 7829 human genomes, we achieve high concordance with population-genetic assumptions and expected inheritance patterns. On simulated data, we show that precision correlates well with our prediction of genotyping difficulty. This together with low memory and time requirements makes our approach well-suited for application in biomedical studies involving small to very large numbers of short-read sequenced genomes.
Source code is available at https://github.com/kehrlab/Complex-SV-Genotyping.
复杂结构变异(SV)是涉及多个 DNA 片段的基因组重排。它们有助于人类多样性,并已被证明会导致孟德尔疾病。然而,我们分析复杂 SV 的能力非常有限。与缺失和其他典型类型的 SV 不同,没有专门为分析复杂 SV 而设计的既定工具。
在这里,我们描述了一种新的计算方法,我们专门为在短读测序基因组中基因分型复杂 SV 而设计。给定变体描述,我们的方法计算基因型特异性的概率分布,用于观察具有广泛属性的对齐读取对。随后,可以使用这些分布来有效地确定在测序基因组中观察到的任何一组对齐读取对的最可能基因型。此外,我们使用这些分布来计算给定变体的基因分型难度,该难度预测实现可靠调用所需的数据量。仔细的评估证实,我们的方法通过在模拟和真实数据中进行可靠的基因型预测,优于其他基因分型器。在多达 7829 个人类基因组上,我们与群体遗传假设和预期的遗传模式高度一致。在模拟数据上,我们表明精度与我们对基因分型难度的预测密切相关。这加上低内存和时间要求,使我们的方法非常适合应用于涉及小到非常大量短读测序基因组的生物医学研究。