Division of Proteome Dynamics, Max Delbrück Center for Molecular Medicine, Berlin, Germany; Faculty of Life Sciences, Humboldt-Universität zu Berlin, Berlin, Germany.
Division of Proteomics, Max Delbrück Center for Molecular Medicine, Berlin, Germany.
Mol Cell Proteomics. 2024 Oct;23(10):100839. doi: 10.1016/j.mcpro.2024.100839. Epub 2024 Sep 11.
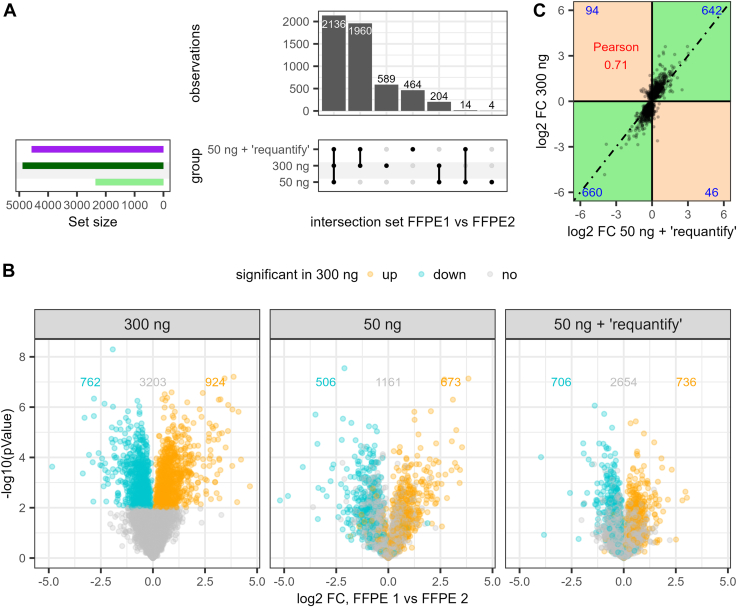
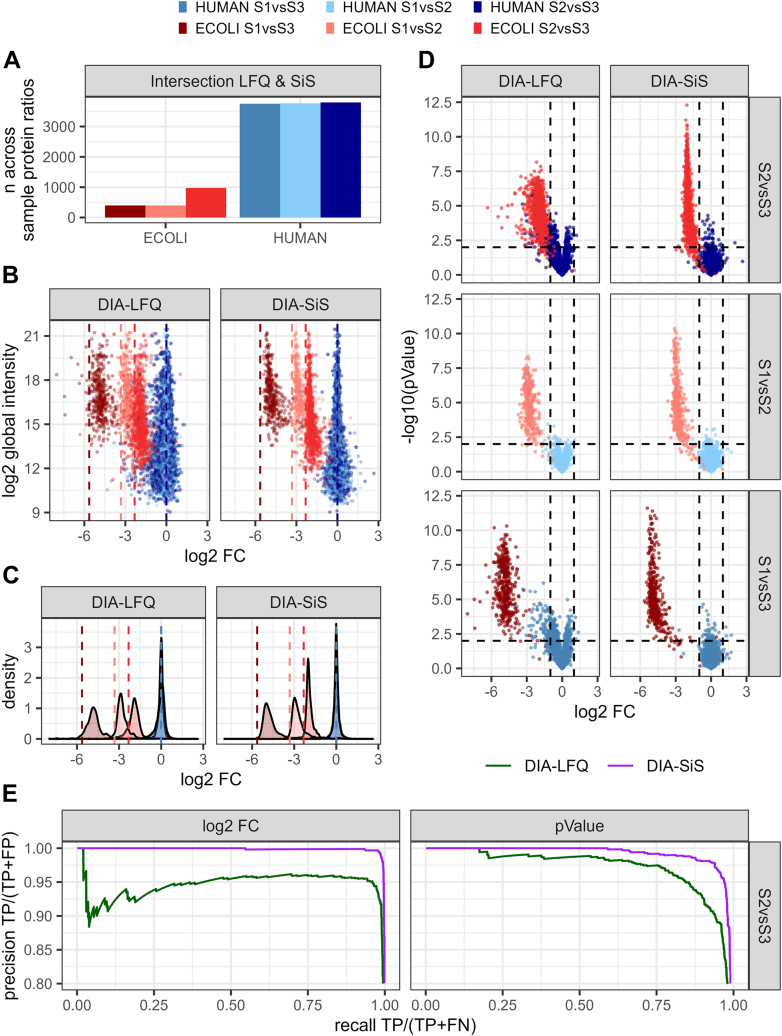
Data-independent acquisition (DIA) is increasingly preferred over data-dependent acquisition due to its higher throughput and fewer missing values. Whereas data-dependent acquisition often uses stable isotope labeling to improve quantification, DIA mostly relies on label-free approaches. Efforts to integrate DIA with isotope labeling include chemical methods like mass differential tags for relative and absolute quantification and dimethyl labeling, which, while effective, complicate sample preparation. Stable isotope labeling by amino acids in cell culture (SILAC) achieves high labeling efficiency through the metabolic incorporation of heavy labels into proteins in vivo. However, the need for metabolic incorporation limits the direct use in clinical scenarios and certain high-throughput experiments. Spike-in SILAC (SiS) methods use an externally generated heavy sample as an internal reference, enabling SILAC-based quantification even for samples that cannot be directly labeled. Here, we combine DIA-SiS, leveraging the robust quantification of SILAC without the complexities associated with chemical labeling. We developed DIA-SiS and rigorously assessed its performance with mixed-species benchmark samples on bulk and single cell-like amount level. We demonstrate that DIA-SiS substantially improves proteome coverage and quantification compared to label-free approaches and reduces incorrectly quantified proteins. Additionally, DIA-SiS proves effective in analyzing proteins in low-input formalin-fixed paraffin-embedded tissue sections. DIA-SiS combines the precision of stable isotope-based quantification with the simplicity of label-free sample preparation, facilitating simple, accurate, and comprehensive proteome profiling.
数据非依赖性采集(DIA)因其高通量和较少的缺失值而越来越受到青睐,优于数据依赖性采集。虽然数据依赖性采集通常使用稳定同位素标记来提高定量准确性,但 DIA 主要依赖于无标记方法。将 DIA 与同位素标记相结合的努力包括化学方法,例如用于相对和绝对定量的质量差分标签和二甲基化标记,虽然有效,但会使样品制备复杂化。稳定同位素标记通过细胞培养中的氨基酸(SILAC)通过在体内将重标记物代谢掺入蛋白质来实现高标记效率。然而,代谢掺入的需求限制了其在临床场景和某些高通量实验中的直接使用。加入 SILAC(SiS)方法使用外部生成的重样品作为内部参考,即使对于无法直接标记的样品,也可以实现基于 SILAC 的定量。在这里,我们结合了 DIA-SiS,利用 SILAC 的强大定量功能,而无需与化学标记相关的复杂性。我们开发了 DIA-SiS,并通过在大量和单细胞样本文库水平上对混合物种基准样品进行严格评估,评估了其性能。我们证明,与无标记方法相比,DIA-SiS 可大大提高蛋白质组覆盖度和定量准确性,并减少错误定量的蛋白质。此外,DIA-SiS 可有效地分析低输入福尔马林固定石蜡包埋组织切片中的蛋白质。DIA-SiS 将稳定同位素定量的精度与无标记样品制备的简单性相结合,促进了简单、准确和全面的蛋白质组分析。