Department of Chemistry , University of Michigan , 930 N. University Avenue, Ann Arbor , Michigan 48109 , United States.
Anal Chem. 2018 Aug 7;90(15):8722-8726. doi: 10.1021/acs.analchem.8b01618. Epub 2018 Jul 19.
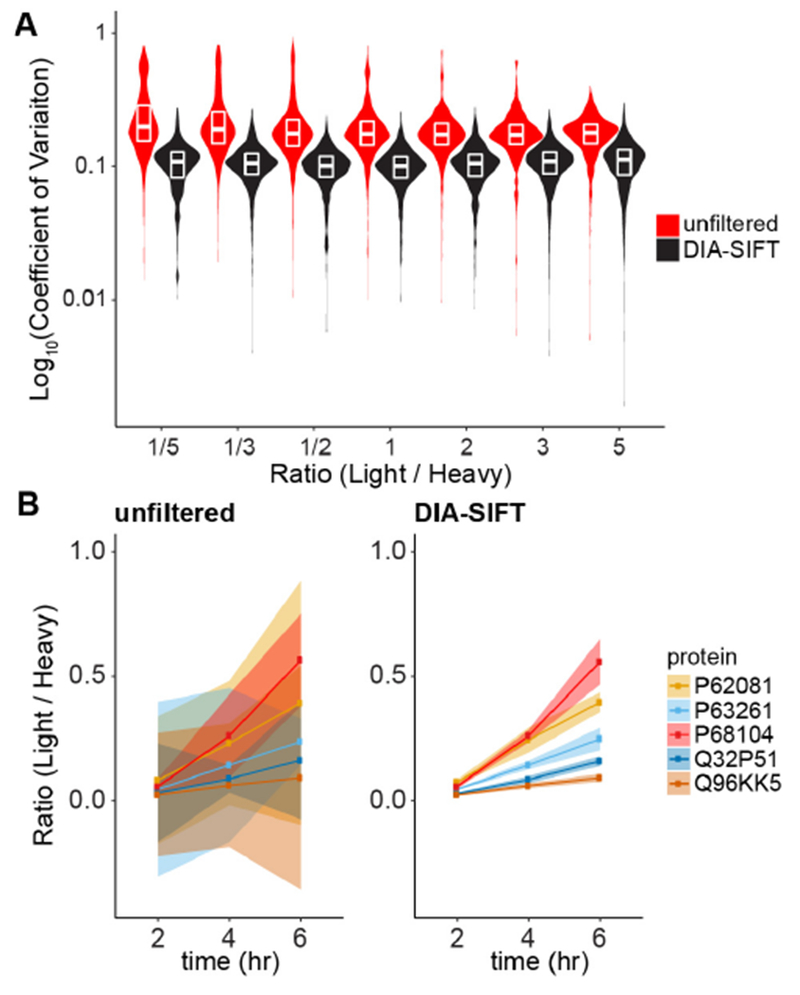
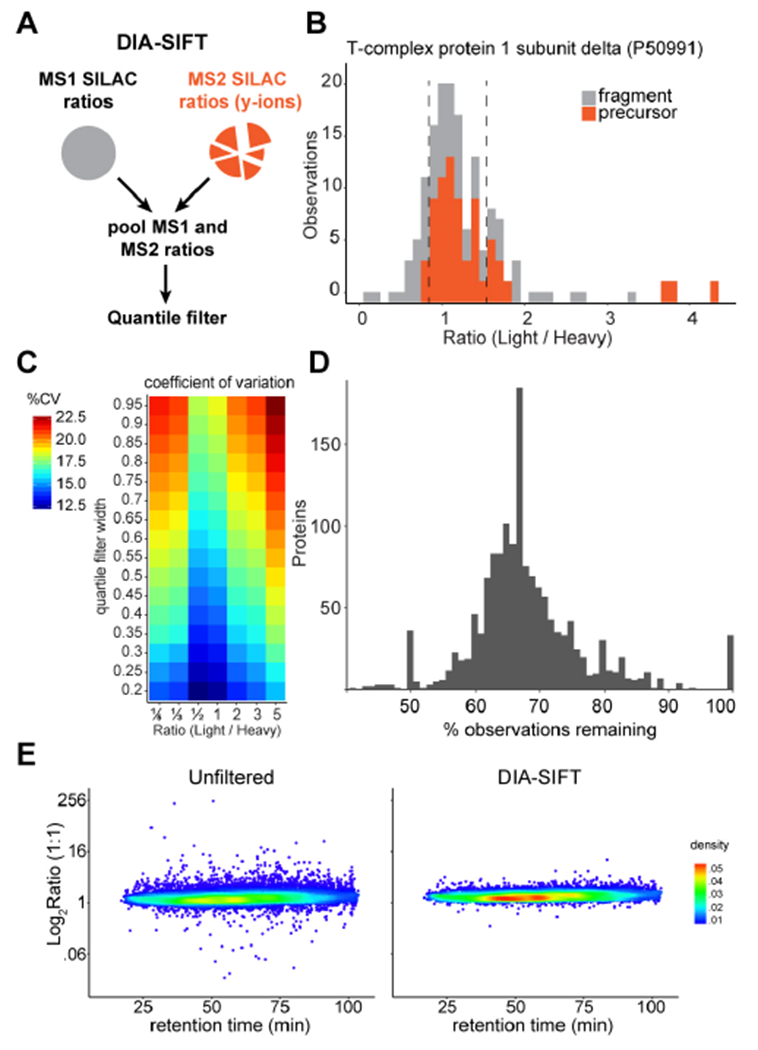
Quantitative mass spectrometry-based protein profiling is widely used to measure protein levels across different treatments or disease states, yet current mass spectrometry acquisition methods present distinct limitations. While data-independent acquisition (DIA) bypasses the stochastic nature of data-dependent acquisition (DDA), fragment spectra derived from DIA are often complex and challenging to deconvolve. In-line ion mobility separation (IMS) adds an additional dimension to increase peak capacity for more efficient product ion assignment. As a similar strategy to sequential window acquisition methods (SWATH), IMS-enabled DIA methods rival DDA methods for protein annotation. Here we evaluate IMS-DIA quantitative accuracy using stable isotope labeling by amino acids in cell culture (SILAC). Since SILAC analysis doubles the sample complexity, we find that IMS-DIA analysis is not sufficiently accurate for sensitive quantitation. However, SILAC precursor pairs share common retention and drift times, and both species cofragment to yield multiple quantifiable isotopic y-ion peak pairs. Since y-ion SILAC ratios are intrinsic for each quantified precursor, combined MS1 and y-ion ratio analysis significantly increases the total number of measurements. With increased sampling, we present DIA-SIFT ( SILAC Intrinsic Filtering Tool), a simple statistical algorithm to identify and eliminate poorly quantified MS1 and/or MS2 events. DIA-SIFT combines both MS1 and y-ion ratios, removes outliers, and provides more accurate and precise quantitation (<15% CV) without removing any proteins from the final analysis. Overall, pooled MS1 and MS2 quantitation increases sampling in IMS-DIA SILAC analyses for accurate and precise quantitation.
基于定量质谱的蛋白质谱分析广泛用于测量不同处理或疾病状态下的蛋白质水平,但目前的质谱采集方法存在明显的局限性。虽然数据非依赖性采集 (DIA) 绕过了数据依赖性采集 (DDA) 的随机性,但 DIA 衍生的片段谱通常很复杂,难以解卷积。在线离子淌度分离 (IMS) 增加了一个额外的维度,以提高峰容量,从而更有效地分配产物离子。作为类似顺序窗口采集方法 (SWATH) 的策略,IMS 增强的 DIA 方法在蛋白质注释方面可与 DDA 方法相媲美。在这里,我们使用稳定同位素标记的细胞培养物中的氨基酸 (SILAC) 来评估 IMS-DIA 的定量准确性。由于 SILAC 分析使样品复杂度增加了一倍,我们发现 IMS-DIA 分析对于敏感定量来说不够准确。然而,SILAC 前体对具有共同的保留和漂移时间,并且两种物质都共同碎裂以产生多个可定量的同位素 y-离子峰对。由于 y-离子 SILAC 比对于每个定量的前体都是内在的,因此结合 MS1 和 y-离子比分析显著增加了总测量数。通过增加采样,我们提出了 DIA-SIFT(SILAC 固有过滤工具),这是一种简单的统计算法,用于识别和消除定量不佳的 MS1 和/或 MS2 事件。DIA-SIFT 结合了 MS1 和 y-离子比,消除了离群值,并提供了更准确和精确的定量(<15%CV),而不会从最终分析中删除任何蛋白质。总的来说,在 IMS-DIA SILAC 分析中合并 MS1 和 MS2 定量可增加采样,以实现准确和精确的定量。