Department of Pediatrics, University of California San Diego, La Jolla, California, USA.
Division of Biological Sciences, University of California San Diego, La Jolla, California, USA.
mSystems. 2024 Oct 22;9(10):e0098524. doi: 10.1128/msystems.00985-24. Epub 2024 Sep 16.
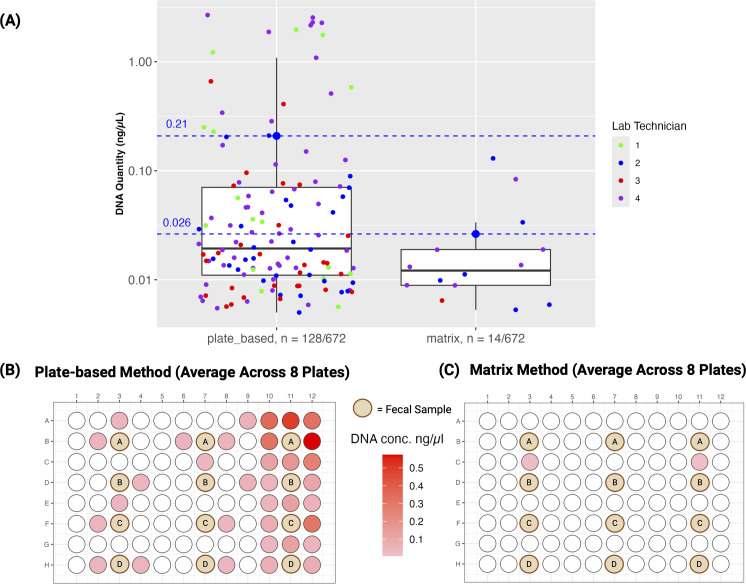
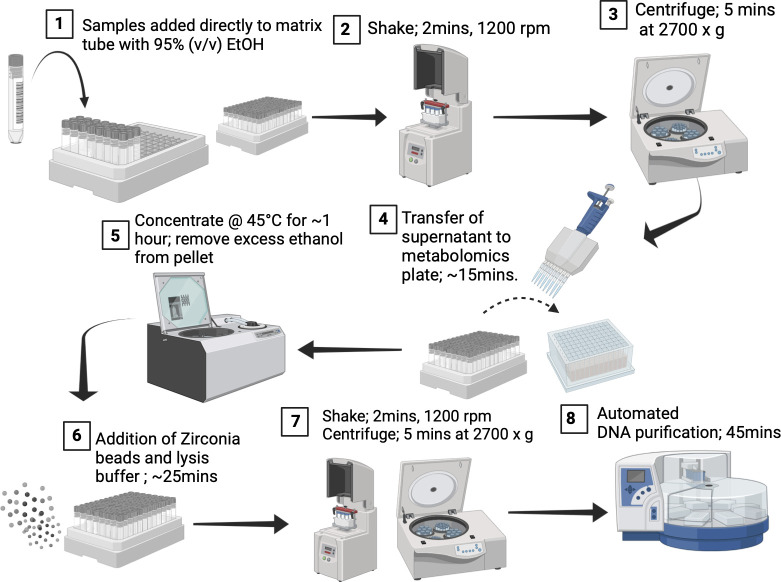
Large-scale studies are essential to answer questions about complex microbial communities that can be extremely dynamic across hosts, environments, and time points. However, managing acquisition, processing, and analysis of large numbers of samples poses many challenges, with cross-contamination being the biggest obstacle. Contamination complicates analysis and results in sample loss, leading to higher costs and constraints on mixed sample type study designs. While many researchers opt for 96-well plates for their workflows, these plates present a significant issue: the shared seal and weak separation between wells leads to well-to-well contamination. To address this concern, we propose an innovative high-throughput approach, termed as the Matrix method, which employs barcoded Matrix Tubes for sample acquisition. This method is complemented by a paired nucleic acid and metabolite extraction, utilizing 95% (vol/vol) ethanol to stabilize microbial communities and as a solvent for extracting metabolites. Comparative analysis between conventional 96-well plate extractions and the Matrix method, measuring 16S rRNA gene levels via quantitative polymerase chain reaction, demonstrates a notable decrease in well-to-well contamination with the Matrix method. Metagenomics, 16S rRNA gene amplicon sequencing (16S), and untargeted metabolomics analysis via liquid chromatography-tandem mass spectrometry (LC-MS/MS) confirmed that the Matrix method recovers reproducible microbial and metabolite compositions that can distinguish between subjects. This advancement is critical for large-scale study design as it minimizes well-to-well contamination and technical variation, shortens processing times, and integrates with automated infrastructure for enhancing sample randomization and metadata generation.
Understanding dynamic microbial communities typically requires large-scale studies. However, handling large numbers of samples introduces many challenges, with cross-contamination being a major issue. It not only complicates analysis but also leads to sample loss and increased costs and restricts diverse study designs. The prevalent use of 96-well plates for nucleic acid and metabolite extractions exacerbates this problem due to their wells having little separation and being connected by a single plate seal. To address this, we propose a new strategy using barcoded Matrix Tubes, showing a significant reduction in cross-contamination compared to conventional plate-based approaches. Additionally, this method facilitates the extraction of both nucleic acids and metabolites from a single tubed sample, eliminating the need to collect separate aliquots for each extraction. This innovation improves large-scale study design by shortening processing times, simplifying analysis, facilitating metadata curation, and producing more reliable results.
大规模研究对于回答有关复杂微生物群落的问题至关重要,这些问题在宿主、环境和时间点上可能极其动态。然而,管理大量样本的获取、处理和分析带来了许多挑战,其中交叉污染是最大的障碍。污染使分析复杂化,并导致样本丢失,从而增加成本并限制混合样本类型的研究设计。尽管许多研究人员选择 96 孔板进行他们的工作流程,但这些板存在一个重大问题:共享密封和孔之间的弱分离导致孔间污染。为了解决这个问题,我们提出了一种创新的高通量方法,称为 Matrix 方法,该方法使用带条形码的 Matrix 管进行样本采集。该方法通过核酸和代谢物提取来补充,使用 95%(体积/体积)乙醇来稳定微生物群落并作为提取代谢物的溶剂。通过定量聚合酶链反应测量 16S rRNA 基因水平,对传统 96 孔板提取与 Matrix 方法进行比较分析,表明 Matrix 方法显著降低了孔间污染。宏基因组学、16S rRNA 基因扩增子测序(16S)和通过液相色谱-串联质谱法(LC-MS/MS)进行的靶向代谢组学分析证实,Matrix 方法可回收可重复的微生物和代谢物组成,可区分研究对象。这项进展对于大规模研究设计至关重要,因为它可以最大限度地减少孔间污染和技术变异,缩短处理时间,并与自动化基础设施集成,以增强样本随机化和元数据生成。
了解动态微生物群落通常需要进行大规模研究。然而,处理大量样本会带来许多挑战,其中交叉污染是一个主要问题。它不仅使分析复杂化,而且还会导致样本丢失,增加成本并限制多样化的研究设计。由于核酸和代谢物提取中使用的 96 孔板的孔之间几乎没有分离,并且由单个板密封连接,因此普遍使用 96 孔板会加剧这个问题。为了解决这个问题,我们提出了一种使用带条形码的 Matrix 管的新策略,与传统的基于板的方法相比,这种方法显著减少了交叉污染。此外,该方法还可以从单个管样中提取核酸和代谢物,无需收集每个提取的单独等分试样。这种创新通过缩短处理时间、简化分析、简化元数据管理以及生成更可靠的结果,改善了大规模研究设计。