The Office of Data Sciences, The Abigail Wexner Research Institute at Nationwide Children's Hospital, 575 Children's Crossroad, Columbus, OH, 43215, USA.
The Steve and Cindy Rasmussen Institute for Genomic Medicine, The Abigail Wexner Research Institute, Nationwide Children's Hospital, 575 Children's Crossroad, Columbus, OH, 43215, USA.
Sci Rep. 2024 Sep 28;14(1):22411. doi: 10.1038/s41598-024-72470-4.
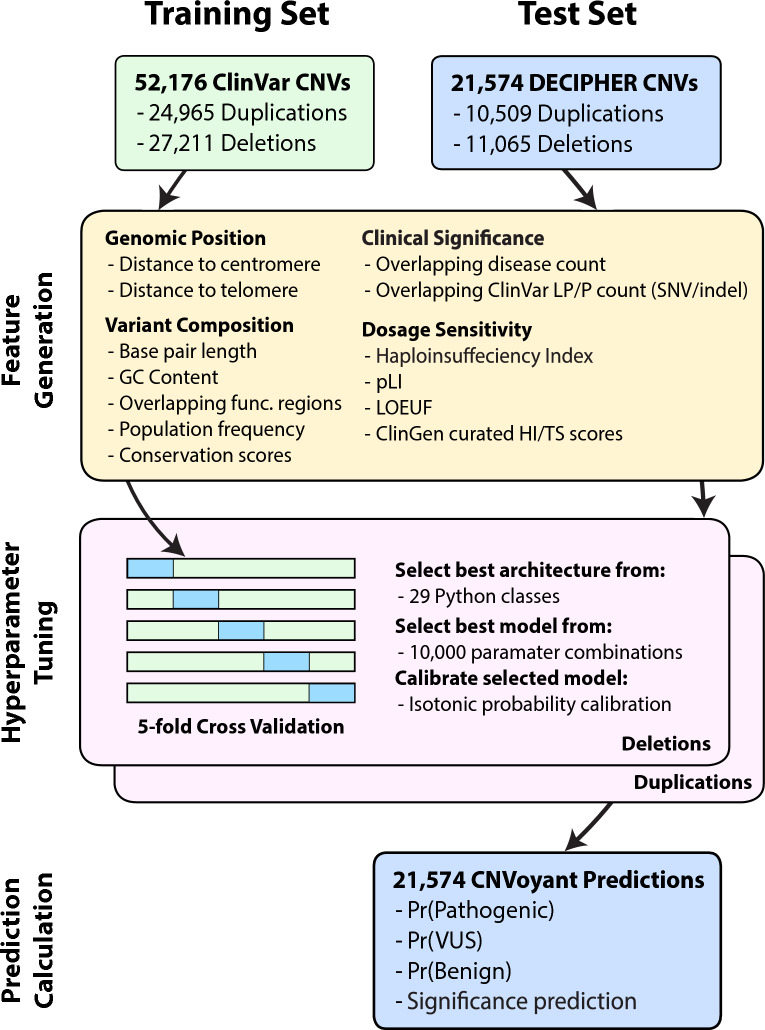
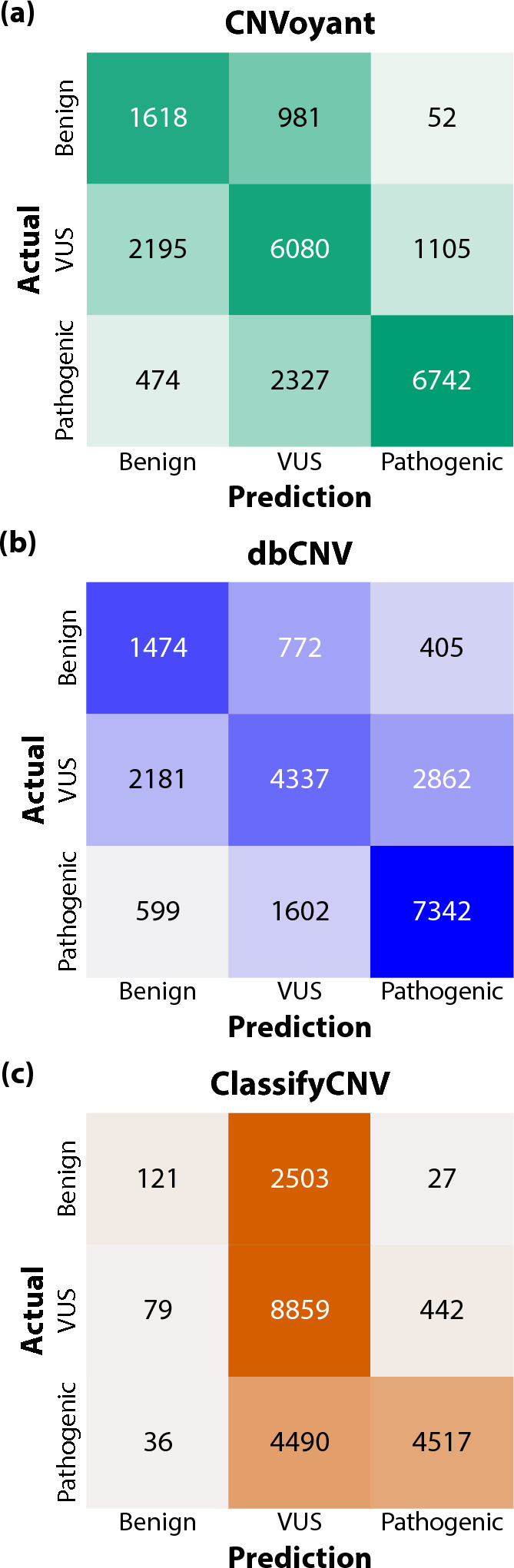
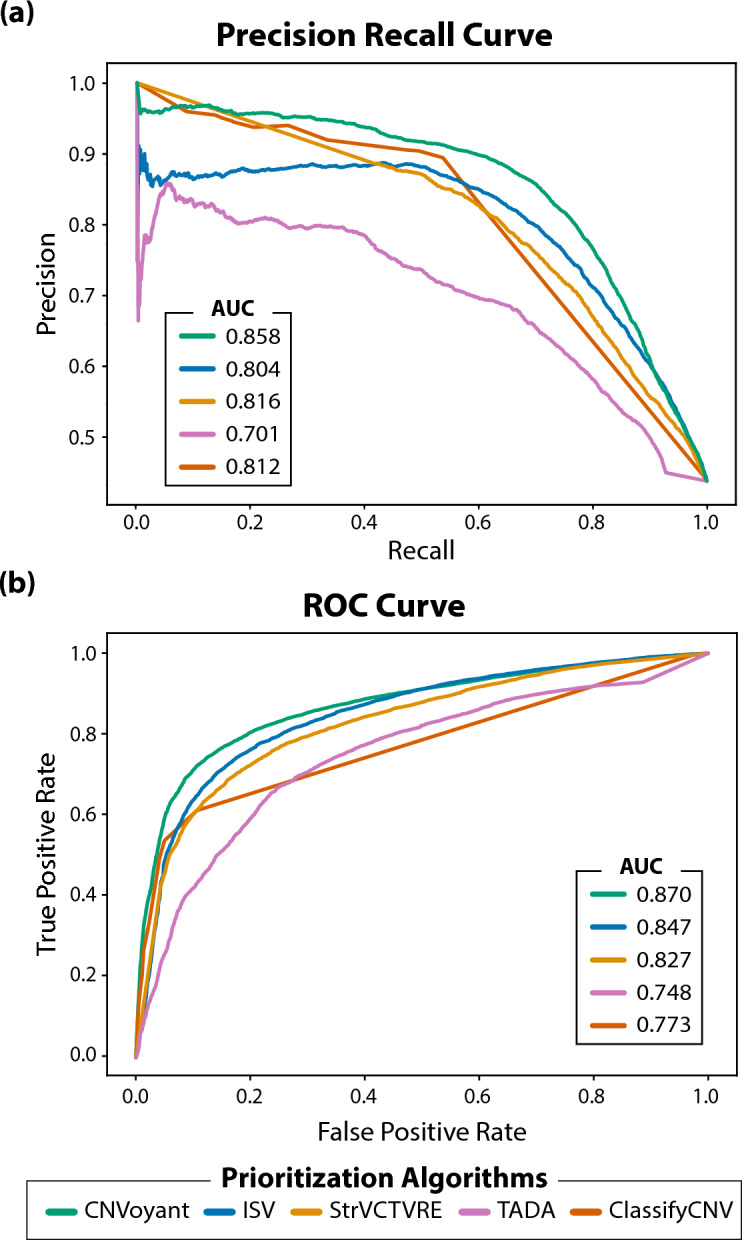
The precise classification of copy number variants (CNVs) presents a significant challenge in genomic medicine, primarily due to the complex nature of CNVs and their diverse impact on rare genetic diseases (RGDs). This complexity is compounded by the limitations of existing methods in accurately distinguishing between benign, uncertain, and pathogenic CNVs. Addressing this gap, we introduce CNVoyant, a machine learning-based multi-class framework designed to enhance the clinical significance classification of CNVs. Trained on a comprehensive dataset of 52,176 ClinVar entries across pathogenic, uncertain, and benign classifications, CNVoyant incorporates a broad spectrum of genomic features, including genome position, disease-gene annotations, dosage sensitivity, and conservation scores. Models to predict the clinical significance of copy number gains and losses were trained independently. Final models were selected after testing 29 machine learning architectures and 10,000 hyperparameter combinations each for deletions and duplications via fivefold cross-validation. We validate the performance of CNVoyant by leveraging a comprehensive set of 21,574 CNVs from the DECIPHER database, a highly regarded resource known for its extensive catalog of chromosomal imbalances linked to clinical outcomes. Compared to alternative approaches, CNVoyant shows marked improvements in precision-recall and ROC AUC metrics for binary pathogenic classifications while going one step further, offering multi-classification of clinical significance and corresponding SHAP explainability plots. Additionally, when provided germline CNV calls from real-world RGD cases with diagnostic CNV(s), CNVoyant correctly classified all diagnostic CNVs as having pathogenic significance with high confidence. This large-scale validation demonstrates CNVoyant's superior accuracy and underscores its potential to aid genomic researchers and clinical geneticists in interpreting the clinical implications of real CNVs.
拷贝数变异 (CNVs) 的精确分类在基因组医学中是一个重大挑战,主要是由于 CNVs 的复杂性及其对罕见遗传疾病 (RGD) 的多种影响。现有的方法在准确区分良性、不确定和致病性 CNVs 方面存在局限性,这使得这种复杂性更加严重。为了解决这一差距,我们引入了 CNVoyant,这是一种基于机器学习的多类框架,旨在增强 CNV 的临床意义分类。该框架在经过致病性、不确定和良性分类的 52,176 个 ClinVar 条目综合数据集上进行了训练,其中包含了广泛的基因组特征,包括基因组位置、疾病基因注释、剂量敏感性和保守分数。分别为拷贝数增益和缺失训练了预测临床意义的模型。通过五重交叉验证,对 29 种机器学习架构和 10,000 种超参数组合进行了测试,然后为缺失和重复分别选择了最终模型。我们通过利用 DECIPHER 数据库中的 21,574 个 CNV 来验证 CNVoyant 的性能,该数据库是一个备受推崇的资源,因其包含与临床结果相关的广泛染色体失衡目录而闻名。与替代方法相比,CNVoyant 在二进制致病性分类的精度-召回率和 ROC AUC 指标方面表现出显著的改进,同时更进一步,提供了临床意义的多分类和相应的 SHAP 可解释性图。此外,当提供来自具有诊断性 CNV 的真实 RGD 病例的种系 CNV 调用时,CNVoyant 以高置信度正确地将所有诊断性 CNV 分类为具有致病性意义。这种大规模验证表明了 CNVoyant 的卓越准确性,并强调了其在帮助基因组研究人员和临床遗传学家解释真实 CNV 的临床意义方面的潜力。