Beer Michael, Oliveira Ana Sofia F, Tooke Catherine L, Hinchliffe Philip, Tsz Yan Li Angie, Balega Balazs, Spencer James, Mulholland Adrian J
School of Cellular and Molecular Medicine, University of Bristol Bristol BS8 1TD UK
Centre for Computational Chemistry, School of Chemistry, University of Bristol BS8 1TS UK
Chem Sci. 2024 Sep 30;15(41):17232-44. doi: 10.1039/d4sc03295k.
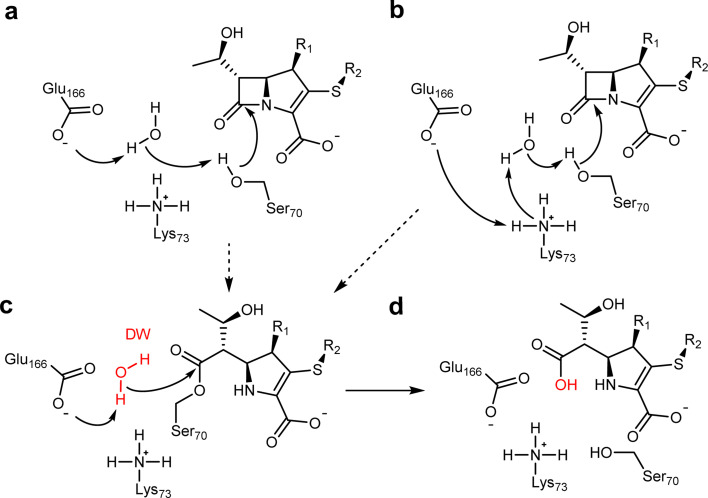
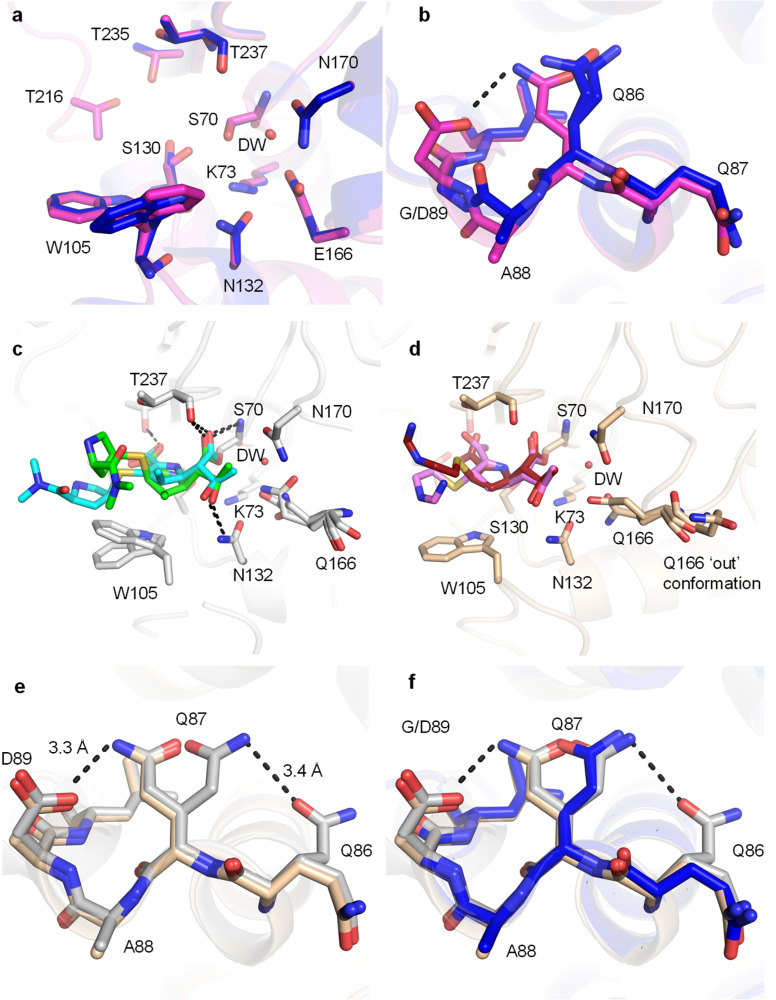
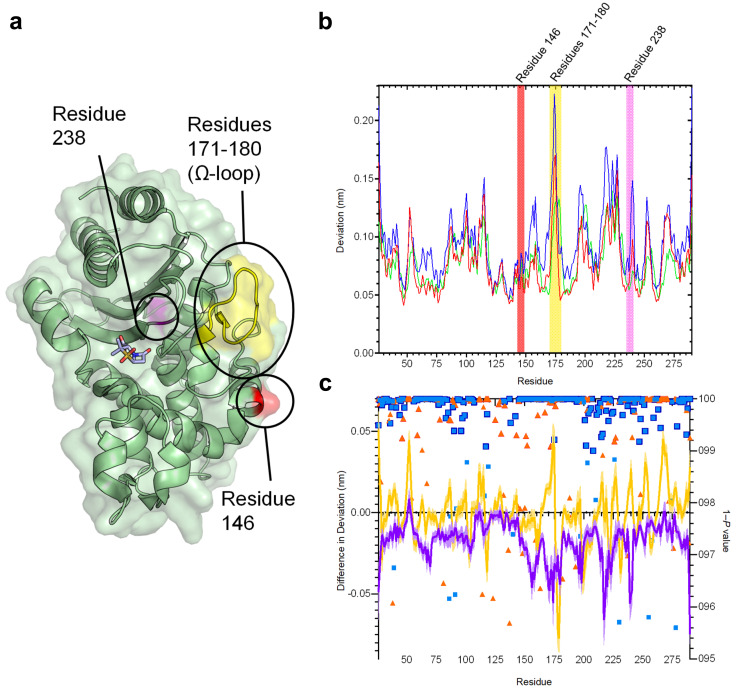
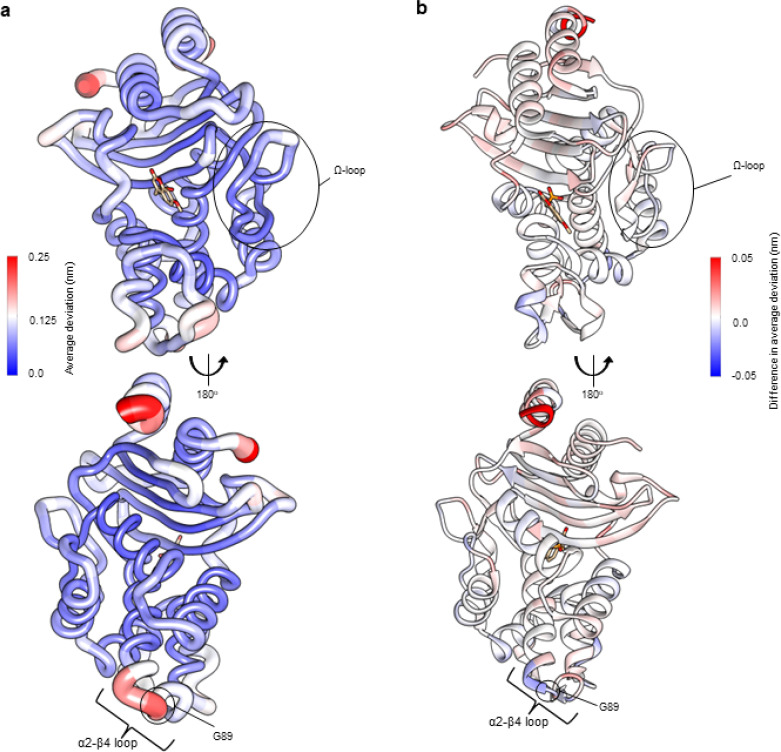
β-Lactamases, which hydrolyse β-lactam antibiotics, are key determinants of antibiotic resistance. Predicting the sites and effects of distal mutations in enzymes is challenging. For β-lactamases, the ability to make such predictions would contribute to understanding activity against, and development of, antibiotics and inhibitors to combat resistance. Here, using dynamical non-equilibrium molecular dynamics (D-NEMD) simulations combined with experiments, we demonstrate that intramolecular communication networks differ in three class A SulpHydryl Variant (SHV)-type β-lactamases. Differences in network architecture and correlated motions link to catalytic efficiency and β-lactam substrate spectrum. Further, the simulations identify a distal residue at position 89 in the clinically important carbapenemase 2 (KPC-2), as a participant in similar networks, suggesting that mutation at this position would modulate enzyme activity. Experimental kinetic, biophysical and structural characterisation of the naturally occurring, but previously biochemically uncharacterised, KPC-2 mutant with several antibiotics and inhibitors reveals significant changes in hydrolytic spectrum, specifically reducing activity towards carbapenems without effecting major structural or stability changes. These results show that D-NEMD simulations can predict distal sites where mutation affects enzyme activity. This approach could have broad application in understanding enzyme evolution, and in engineering of natural and enzymes.
β-内酰胺酶可水解β-内酰胺类抗生素,是抗生素耐药性的关键决定因素。预测酶中远距离突变的位点和影响具有挑战性。对于β-内酰胺酶而言,进行此类预测的能力将有助于理解其对抗生素的活性以及对抗耐药性的抗生素和抑制剂的开发。在此,我们结合实验使用动态非平衡分子动力学(D-NEMD)模拟,证明了三种A类巯基变体(SHV)型β-内酰胺酶的分子内通信网络存在差异。网络结构和相关运动的差异与催化效率和β-内酰胺底物谱相关。此外,模拟确定了临床上重要的碳青霉烯酶2(KPC-2)中第89位的一个远距离残基参与了类似的网络,这表明该位置的突变会调节酶的活性。对天然存在但以前未经生化表征的KPC-2突变体与几种抗生素和抑制剂进行的实验动力学、生物物理和结构表征揭示了水解谱的显著变化,特别是降低了对碳青霉烯类的活性,而没有影响主要的结构或稳定性变化。这些结果表明,D-NEMD模拟可以预测突变影响酶活性的远距离位点。这种方法在理解酶的进化以及天然酶和工程酶的设计方面可能具有广泛的应用。