Dipartimento di Medicina Sperimentale, Università degli Studi di Genova, Genova, Italia.
Present Affiliation: Department of Cell Biology, Universidad de Granada, Granada, Spain.
Cell Mol Life Sci. 2024 Oct 5;81(1):416. doi: 10.1007/s00018-024-05441-7.
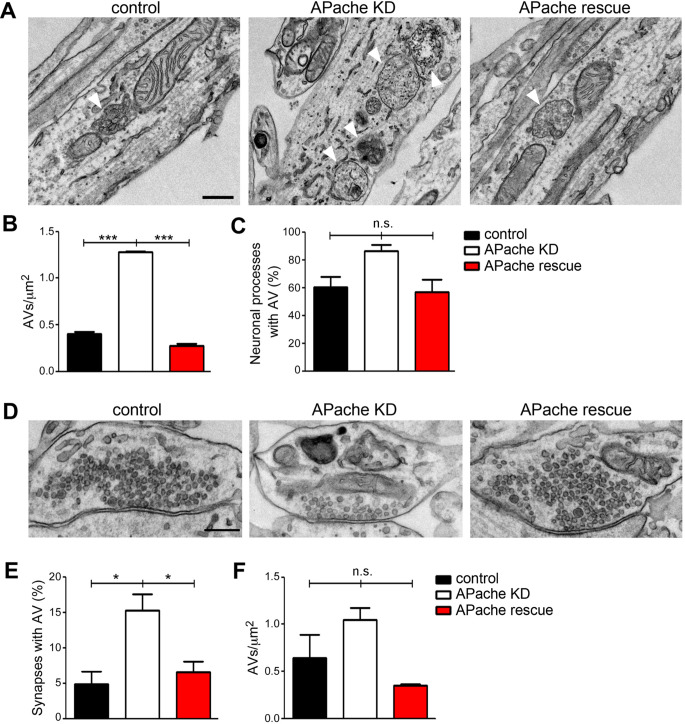
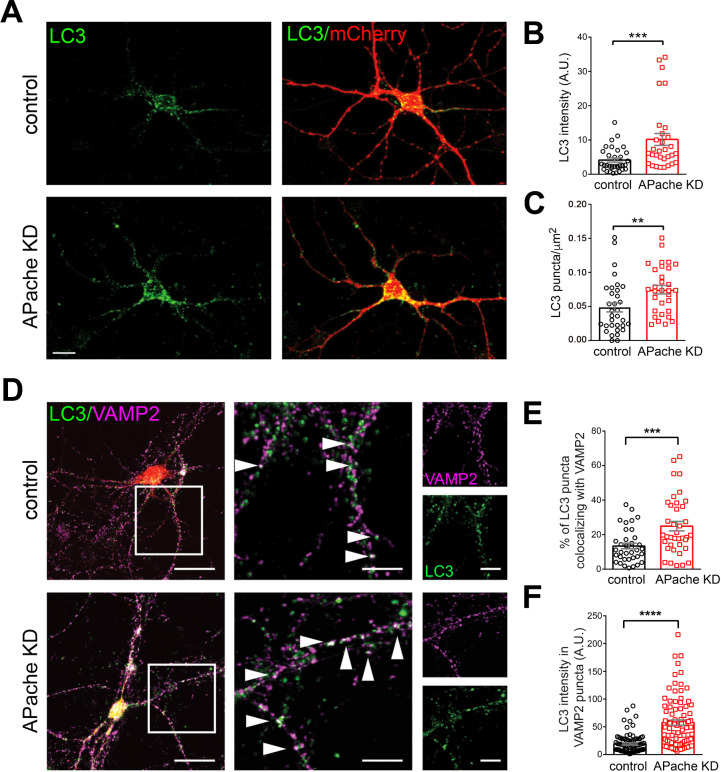
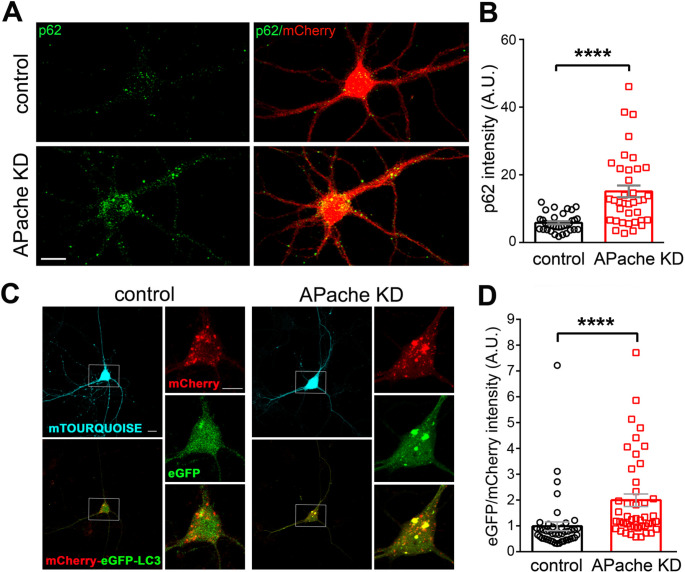
Neurons are dependent on efficient quality control mechanisms to maintain cellular homeostasis and function due to their polarization and long-life span. Autophagy is a lysosomal degradative pathway that provides nutrients during starvation and recycles damaged and/or aged proteins and organelles. In neurons, autophagosomes constitutively form in distal axons and at synapses and are trafficked retrogradely to the cell soma to fuse with lysosomes for cargo degradation. How the neuronal autophagy pathway is organized and controlled remains poorly understood. Several presynaptic endocytic proteins have been shown to regulate both synaptic vesicle recycling and autophagy. Here, by combining electron, fluorescence, and live imaging microscopy with biochemical analysis, we show that the neuron-specific protein APache, a presynaptic AP-2 interactor, functions in neurons as an important player in the autophagy process, regulating the retrograde transport of autophagosomes. We found that APache colocalizes and co-traffics with autophagosomes in primary cortical neurons and that induction of autophagy by mTOR inhibition increases LC3 and APache protein levels at synaptic boutons. APache silencing causes a blockade of autophagic flux preventing the clearance of p62/SQSTM1, leading to a severe accumulation of autophagosomes and amphisomes at synaptic terminals and along neurites due to defective retrograde transport of TrkB-containing signaling amphisomes along the axons. Together, our data identify APache as a regulator of the autophagic cycle, potentially in cooperation with AP-2, and hypothesize that its dysfunctions contribute to the early synaptic impairments in neurodegenerative conditions associated with impaired autophagy.
神经元由于其极化和长寿命而依赖于有效的质量控制机制来维持细胞内稳态和功能。自噬是一种溶酶体降解途径,可在饥饿时提供营养,并回收受损和/或衰老的蛋白质和细胞器。在神经元中,自噬体在远端轴突和突触处持续形成,并逆行运输到细胞体与溶酶体融合以降解货物。神经元自噬途径如何组织和控制仍知之甚少。已经表明几种突触前内吞蛋白可调节突触小泡再循环和自噬。在这里,我们通过结合电子、荧光和活细胞成像显微镜与生化分析,表明神经元特异性蛋白 APache,一种突触前 AP-2 相互作用蛋白,在神经元中作为自噬过程中的重要参与者发挥作用,调节自噬体的逆行运输。我们发现 APache 在原代皮质神经元中与自噬体共定位和共运输,并且 mTOR 抑制诱导的自噬增加突触小体处的 LC3 和 APache 蛋白水平。APache 沉默会阻止自噬流,防止 p62/SQSTM1 的清除,从而导致由于 TrkB 包含的信号转导内体沿轴突的逆行运输缺陷,突触末端和神经突沿线的自噬体和内体严重积累。总之,我们的数据将 APache 鉴定为自噬循环的调节剂,可能与 AP-2 合作,并假设其功能障碍导致与自噬受损相关的神经退行性疾病中的早期突触损伤。