Bioinformatics Lab, Advanced Research Institute for Informatics, Computing, and Networking, De La Salle University Manila, 2401 Taft Avenue, Manila, Philippines.
Department of Software Technology, College of Computer Studies, De La Salle University Manila, 2401 Taft Avenue, Manila, Philippines.
BMC Bioinformatics. 2024 Oct 24;25(Suppl 2):335. doi: 10.1186/s12859-024-05924-1.
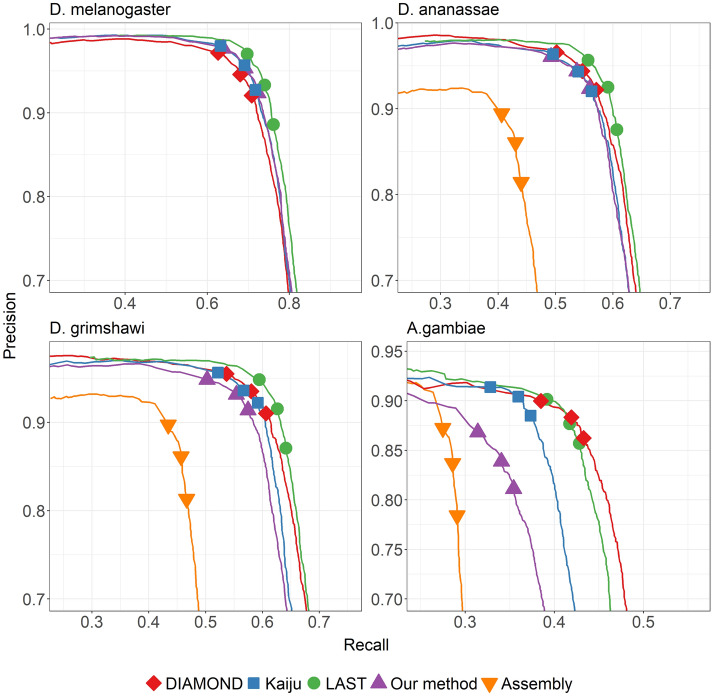
Conventional differential gene expression analysis pipelines for non-model organisms require computationally expensive transcriptome assembly. We recently proposed an alternative strategy of directly aligning RNA-seq reads to a protein database, and demonstrated drastic improvements in speed, memory usage, and accuracy in identifying differentially expressed genes.
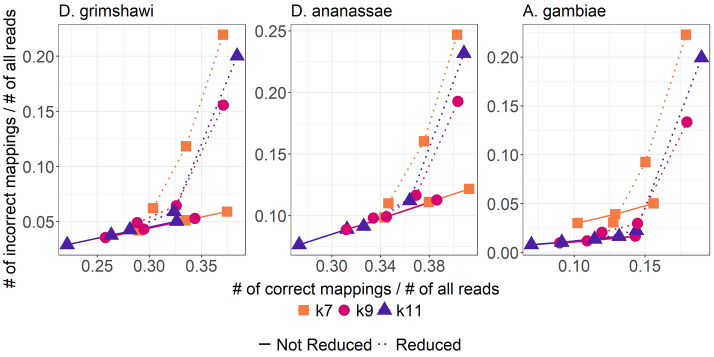
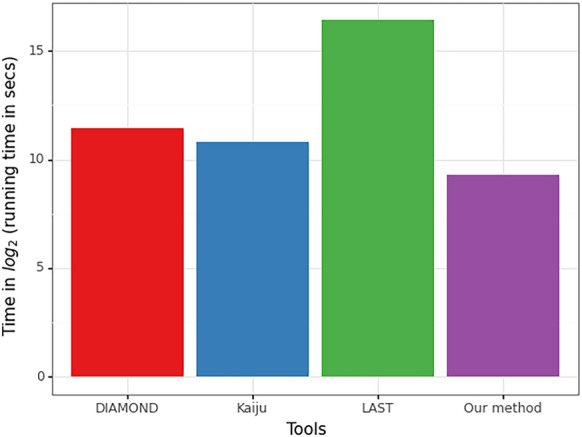
Here we report a further speed-up by replacing DNA-protein alignment by quasi-mapping, making our pipeline > 1000× faster than assembly-based approach, and still more accurate. We also compare quasi-mapping to other mapping techniques, and show that it is faster but at the cost of sensitivity.
We provide a quick-and-dirty differential gene expression analysis pipeline for non-model organisms without a reference transcriptome, which directly quasi-maps RNA-seq reads to a reference protein database, avoiding computationally expensive transcriptome assembly.
针对非模式生物的常规差异基因表达分析流程需要进行计算成本高昂的转录组组装。我们最近提出了一种替代策略,即将 RNA-seq reads 直接比对到蛋白质数据库,在识别差异表达基因方面,该策略在速度、内存使用和准确性方面均有显著提高。
在这里,我们通过用准映射替代 DNA-蛋白质比对,进一步提高了速度,使我们的流程比基于组装的方法快 1000 多倍,且准确性更高。我们还将准映射与其他映射技术进行了比较,并表明它速度更快,但代价是敏感性降低。
我们为没有参考转录组的非模式生物提供了一种快速而粗略的差异基因表达分析流程,该流程直接将 RNA-seq 读取准映射到参考蛋白质数据库,避免了计算成本高昂的转录组组装。