Vock Isaac W, Mabin Justin W, Machyna Martin, Zhang Alexandra, Hogg J Robert, Simon Matthew D
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut 06520, USA.
Institute of Biomolecular Design and Discovery, Yale University, West Haven, Connecticut 06516, USA.
bioRxiv. 2024 Oct 17:2024.10.14.617411. doi: 10.1101/2024.10.14.617411.
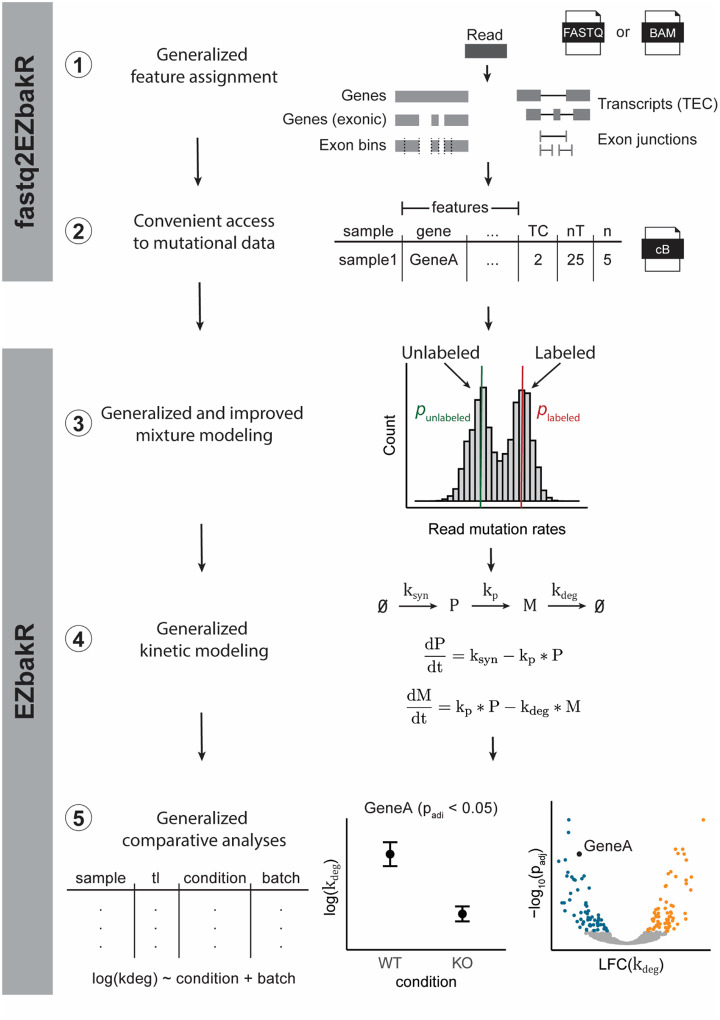
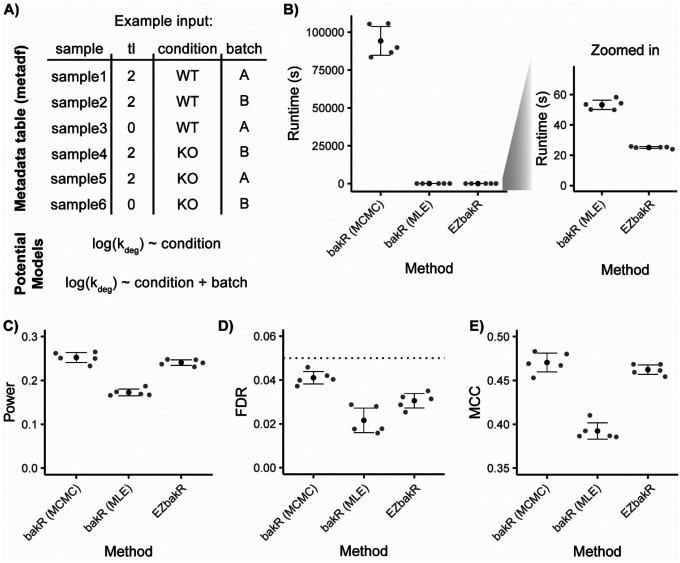
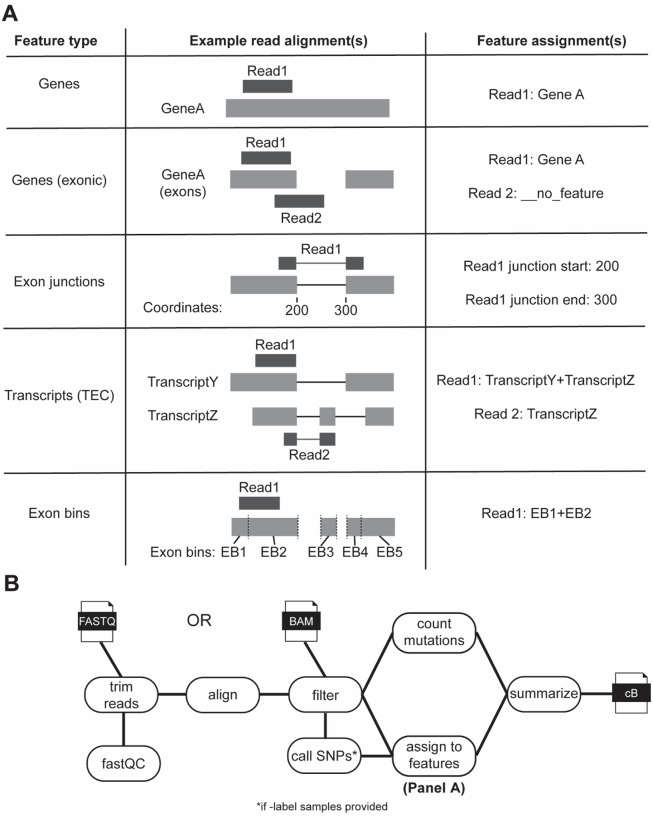
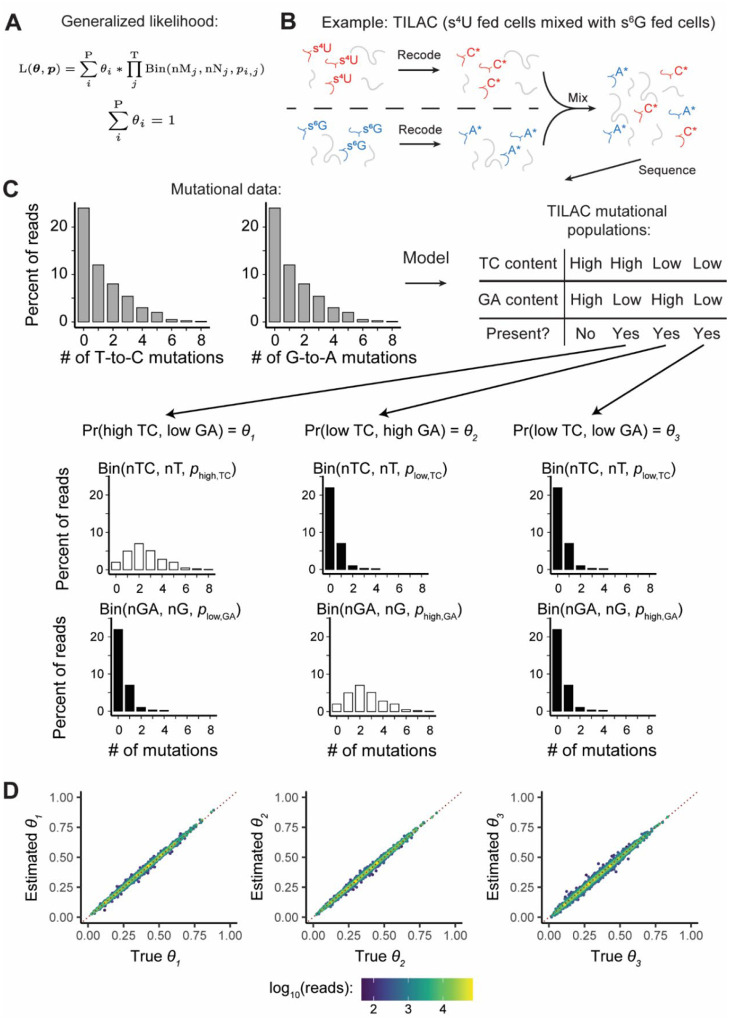
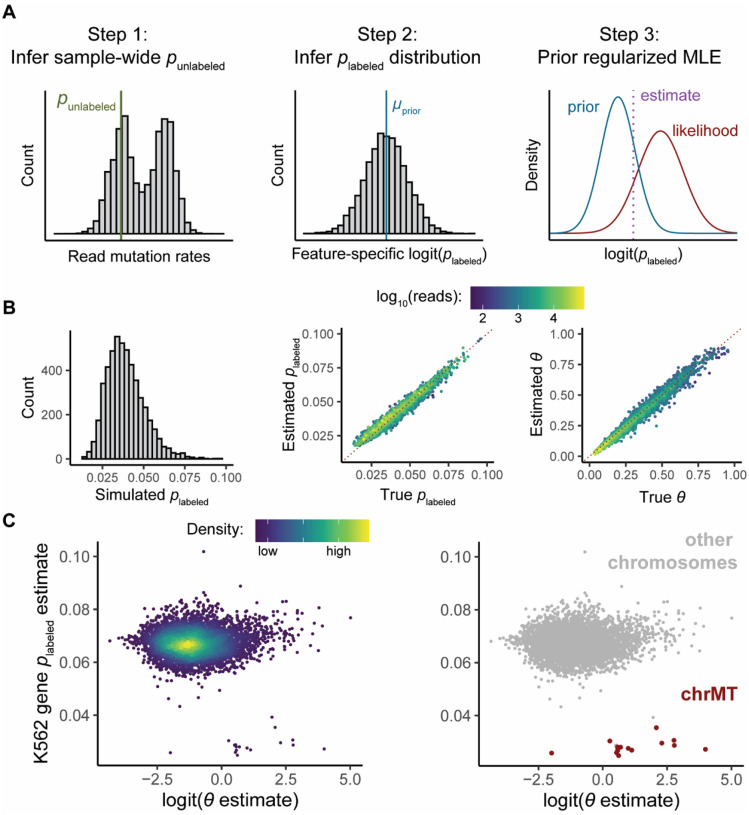
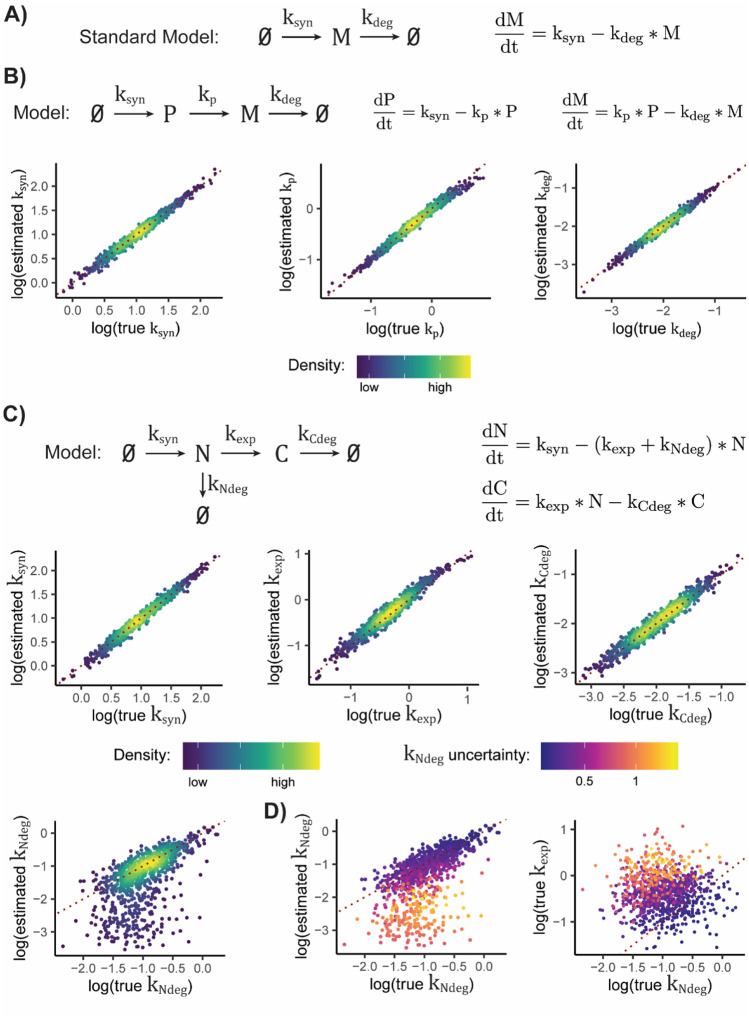
Nucleotide recoding RNA sequencing methods (NR-seq; TimeLapse-seq, SLAM-seq, TUC-seq, etc.) are powerful approaches for assaying transcript population dynamics. In addition, these methods have been extended to probe a host of regulated steps in the RNA life cycle. Current bioinformatic tools significantly constrain analyses of NR-seq data. To address this limitation, we developed EZbakR, an R package to facilitate a more comprehensive set of NR-seq analyses, and fastq2EZbakR, a Snakemake pipeline for flexible preprocessing of NR-seq datasets, collectively referred to as the EZbakR suite. Together, these tools generalize many aspects of the NR-seq analysis workflow. The fastq2EZbakR pipeline can assign reads to a diverse set of genomic features (e.g., genes, exons, splice junctions, etc.), and EZbakR can perform analyses on any combination of these features. EZbakR extends standard NR-seq mutational modeling to support multi-label analyses (e.g., sU and sG dual labeling), and implements an improved hierarchical model to better account for transcript-to-transcript variance in metabolic label incorporation. EZbakR also generalizes dynamical systems modeling of NR-seq data to support analyses of premature mRNA processing and flow between subcellular compartments. Finally, EZbakR implements flexible and well-powered comparative analyses of all estimated parameters via design matrix-specified generalized linear modeling. The EZbakR suite will thus allow researchers to make full, effective use of NR-seq data.
核苷酸重编码RNA测序方法(NR-seq;TimeLapse-seq、SLAM-seq、TUC-seq等)是用于分析转录本群体动态的强大方法。此外,这些方法已被扩展用于探究RNA生命周期中一系列受调控的步骤。当前的生物信息学工具显著限制了对NR-seq数据的分析。为解决这一局限性,我们开发了EZbakR(一个R包,以促进更全面的NR-seq分析集)和fastq2EZbakR(一个用于灵活预处理NR-seq数据集的Snakemake管道),统称为EZbakR套件。这些工具共同概括了NR-seq分析工作流程的许多方面。fastq2EZbakR管道可以将 reads 分配到各种基因组特征(例如基因、外显子、剪接位点等),并且EZbakR可以对这些特征的任何组合进行分析。EZbakR扩展了标准的NR-seq突变建模以支持多标签分析(例如sU和sG双标记),并实现了一种改进的层次模型以更好地考虑代谢标签掺入中的转录本间差异。EZbakR还概括了NR-seq数据的动态系统建模,以支持对过早mRNA加工和亚细胞区室间流动的分析。最后,EZbakR通过设计矩阵指定的广义线性建模对所有估计参数进行灵活且强大的比较分析。因此,EZbakR套件将使研究人员能够充分、有效地利用NR-seq数据。