Labcorp Genetics Inc (formerly Invitae Corporation), San Francisco, California.
Invitae Corporation (now part of Labcorp Genetics), San Francisco, California.
JAMA Netw Open. 2024 Nov 4;7(11):e2444526. doi: 10.1001/jamanetworkopen.2024.44526.
Because accurate and consistent classification of DNA sequence variants is fundamental to germline genetic testing, understanding patterns of initial variant classification (VC) and subsequent reclassification from large-scale, empirical data can help improve VC methods, promote equity among race, ethnicity, and ancestry (REA) groups, and provide insights to inform clinical practice.
To measure the degree to which initial VCs met certainty thresholds set by professional guidelines and quantify the rates of, the factors associated with, and the impact of reclassification among more than 2 million variants.
DESIGN, SETTING, AND PARTICIPANTS: This cohort study used clinical multigene panel and exome sequencing data from diagnostic testing for hereditary disorders, carrier screening, or preventive genetic screening from individuals for whom genetic testing was performed between January 1, 2015, and June 30, 2023.
DNA variants were classified into 1 of 5 categories: benign, likely benign, variant of uncertain significance (VUS), likely pathogenic, or pathogenic.
The main outcomes were accuracy of classifications, rates and directions of reclassifications, evidence contributing to reclassifications, and their impact across different clinical areas and REA groups. One-way analysis of variance followed by post hoc pairwise Tukey honest significant difference tests were used to analyze differences among means, and pairwise Pearson χ2 tests with Bonferroni corrections were used to compare categorical variables among groups.
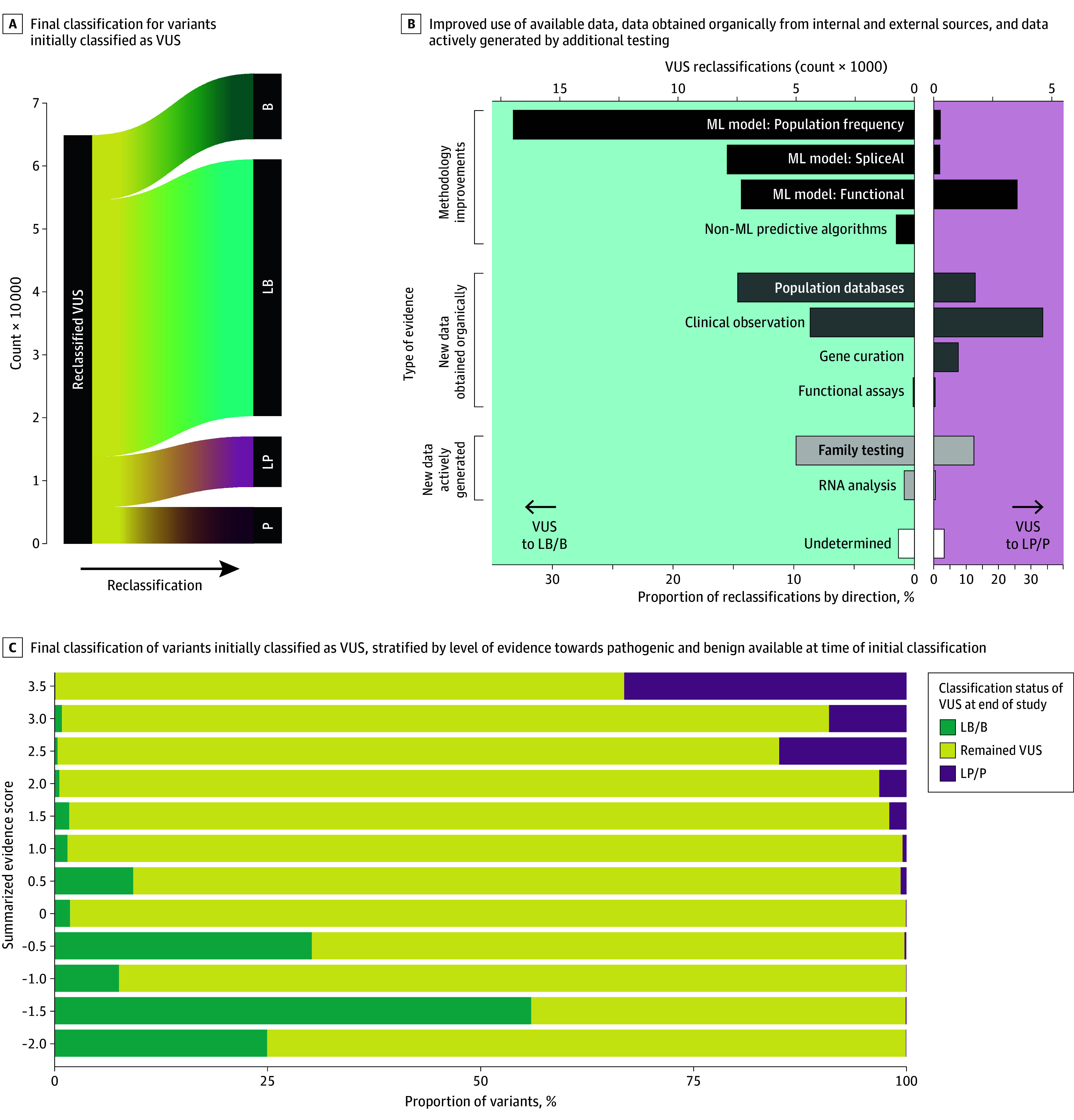
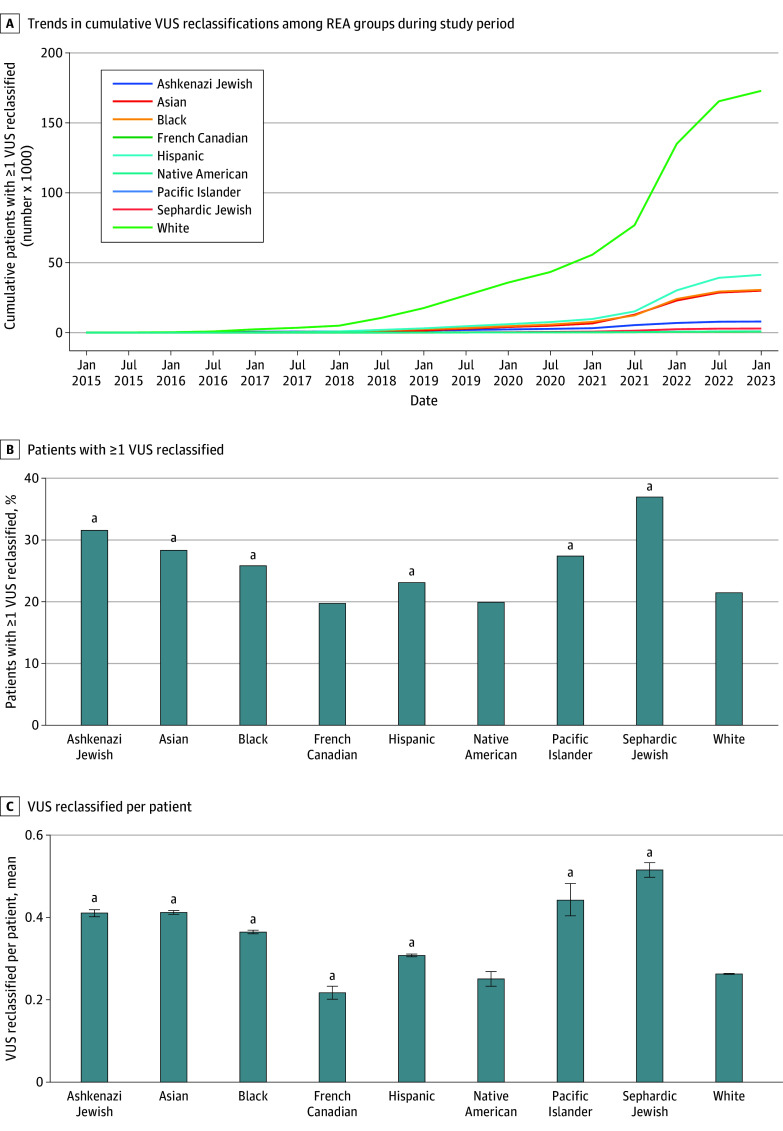
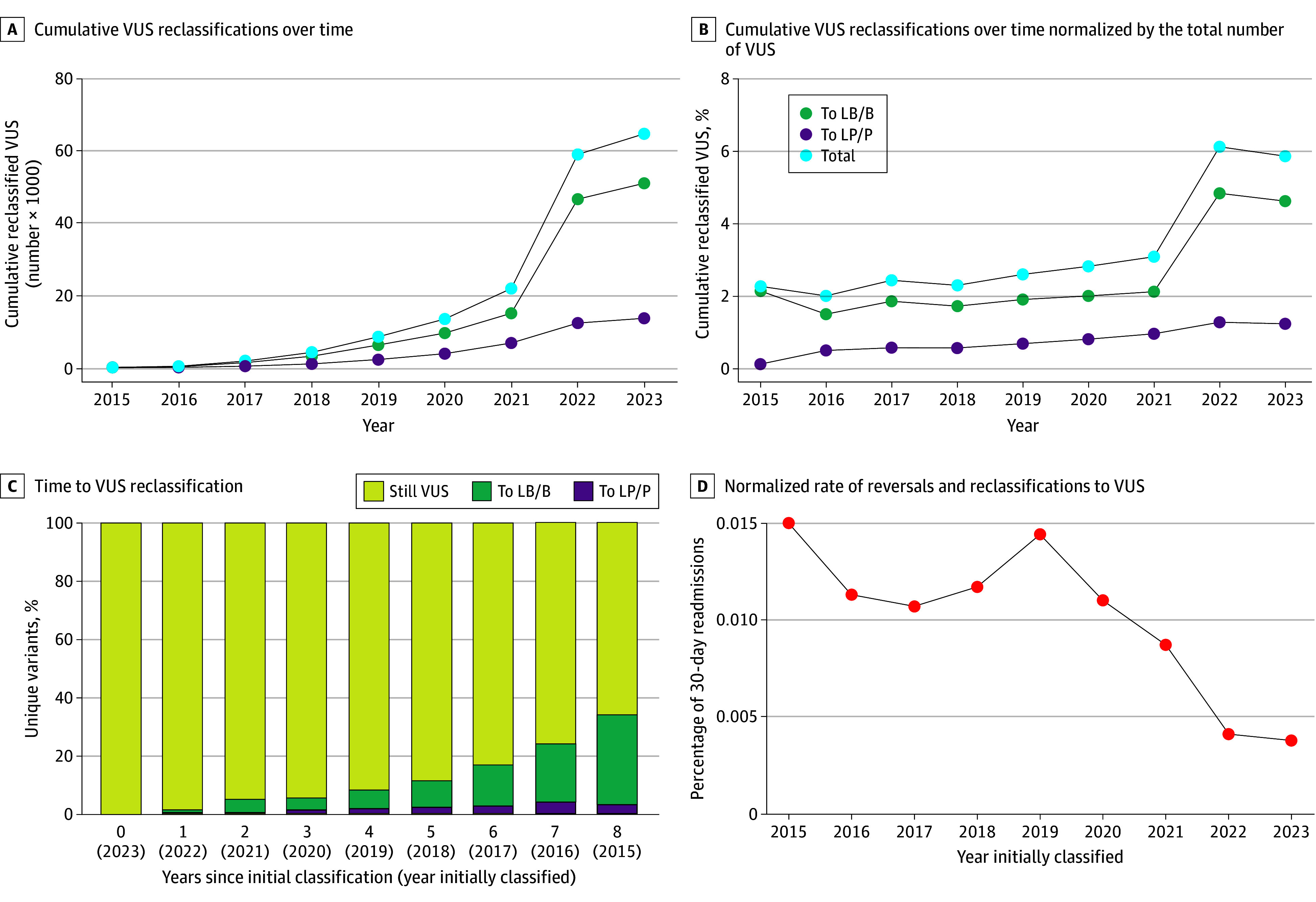
The cohort comprised 3 272 035 individuals (median [range] age, 44 [0-89] years; 2 240 506 female [68.47%] and 1 030 729 male [31.50%]; 216 752 Black [6.62%]; 336 414 Hispanic [10.28%]; 1 804 273 White [55.14%]). Among 2 051 736 variants observed over 8 years in this cohort, 94 453 (4.60%) were reclassified. Some variants were reclassified more than once, resulting in 105 172 total reclassification events. The majority (64 752 events [61.65%]) were changes from VUS to either likely benign, benign, likely pathogenic, or pathogenic categories. An additional 37.66% of reclassifications (39 608 events) were gains in classification certainty to terminal categories (ie, likely benign to benign and likely pathogenic to pathogenic). Only a small fraction (663 events [0.63%]) moved toward less certainty, or very rarely (61 events [0.06%]) were classification reversals. When normalized by the number of individuals tested, VUS reclassification rates were higher among specific underrepresented REA populations (Ashkenazi Jewish, Asian, Black, Hispanic, Pacific Islander, and Sephardic Jewish). Approximately one-half of VUS reclassifications (37 074 of 64 840 events [57.18%]) resulted from improved use of data from computational modeling.
In this cohort study of individuals undergoing genetic testing, the empirically estimated accuracy of pathogenic, likely pathogenic, benign, and likely benign classifications exceeded the certainty thresholds set by current VC guidelines, suggesting the need to reevaluate definitions of these classifications. The relative contribution of various strategies to resolve VUS, including emerging machine learning-based computational methods, RNA analysis, and cascade family testing, provides useful insights that can be applied toward further improving VC methods, reducing the rate of VUS, and generating more definitive results for patients.
由于准确且一致的 DNA 序列变异分类是生殖系基因检测的基础,因此了解从大型经验数据初始变异分类 (VC) 和后续重新分类的模式,可以帮助改进 VC 方法,促进种族、民族和血统 (REA) 群体之间的公平,并提供信息以指导临床实践。
衡量初始 VC 符合专业指南设定的确定性阈值的程度,并量化超过 200 万个变体的重新分类率、与重新分类相关的因素以及重新分类的影响。
设计、设置和参与者:这项队列研究使用了来自诊断性遗传疾病检测、携带者筛查或预防性基因筛查的临床多基因面板和外显子测序数据,这些数据来自于 2015 年 1 月 1 日至 2023 年 6 月 30 日期间进行基因检测的个体。
DNA 变体被分类为 5 个类别之一:良性、可能良性、意义不明的变异 (VUS)、可能致病性或致病性。
主要结果是分类的准确性、重新分类的率和方向、导致重新分类的证据,以及它们在不同临床领域和 REA 群体中的影响。采用单因素方差分析,然后是事后两两 Tukey 诚实显著差异检验分析均值之间的差异,采用两两 Pearson χ2 检验和 Bonferroni 校正分析组间的分类变量。
该队列包括 3272035 名个体(中位数[范围]年龄,44[0-89]岁;2240506 名女性[68.47%]和 1030729 名男性[31.50%];216752 名黑人[6.62%];336414 名西班牙裔[10.28%];1804273 名白人[55.14%])。在该队列 8 年的观察中,2051736 个变体中有 94453 个(4.60%)被重新分类。有些变体被重新分类多次,导致总共 105172 个重新分类事件。大多数(64752 个事件[61.65%])是从 VUS 变为良性、可能良性、可能致病性或致病性类别。另外 37.66%(39608 个事件)的重新分类是将分类确定性提高到终末类别(即从可能良性变为良性和可能致病性变为致病性)。只有一小部分(663 个事件[0.63%])朝着不太确定的方向移动,或者很少(61 个事件[0.06%])是分类逆转。按受检个体数归一化后,特定代表性不足的 REA 人群(阿什肯纳兹犹太裔、亚裔、黑人、西班牙裔、太平洋岛民和塞法迪犹太裔)的 VUS 重新分类率较高。大约一半的 VUS 重新分类(64840 个事件中的 37074 个[57.18%])是由于更好地利用了计算建模数据。
在这项对接受基因检测的个体进行的队列研究中,致病性、可能致病性、良性和可能良性分类的经验估计准确性超过了当前 VC 指南设定的确定性阈值,这表明需要重新评估这些分类的定义。各种策略(包括新兴的基于机器学习的计算方法、RNA 分析和级联家族测试)对解决 VUS 的相对贡献提供了有用的见解,可应用于进一步改进 VC 方法、降低 VUS 率,并为患者生成更明确的结果。