Gluth Austin, Li Xiaolu, Gritsenko Marina A, Gaffrey Matthew J, Kim Doo Nam, Lalli Priscila M, Chu Rosalie K, Day Nicholas J, Sagendorf Tyler J, Monroe Matthew E, Feng Song, Liu Tao, Yang Bin, Qian Wei-Jun, Zhang Tong
Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington, USA; Department of Biological Systems Engineering, Washington State University, Richland, Washington, USA.
Biological Sciences Division, Pacific Northwest National Laboratory, Richland, Washington, USA.
Mol Cell Proteomics. 2024 Dec;23(12):100881. doi: 10.1016/j.mcpro.2024.100881. Epub 2024 Nov 15.
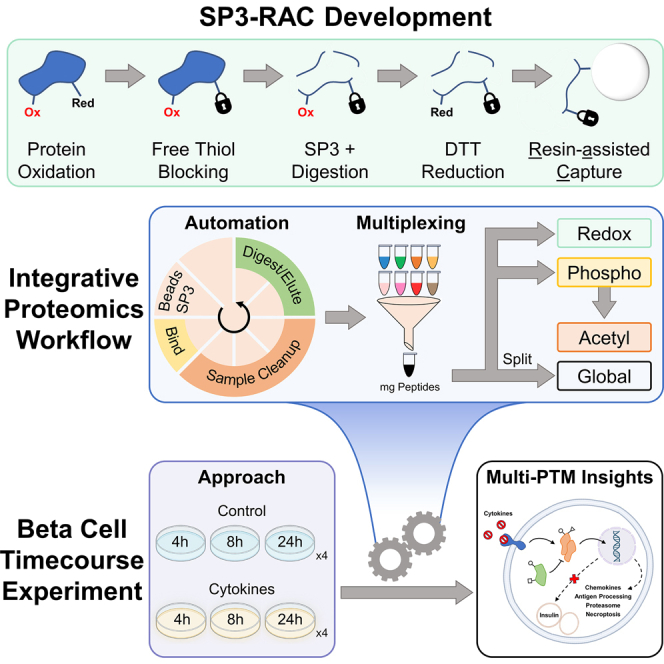
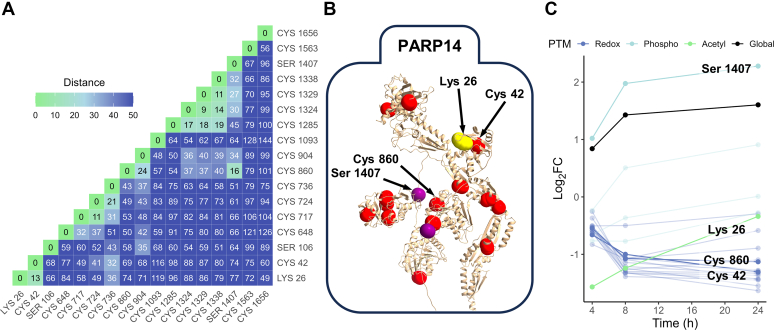
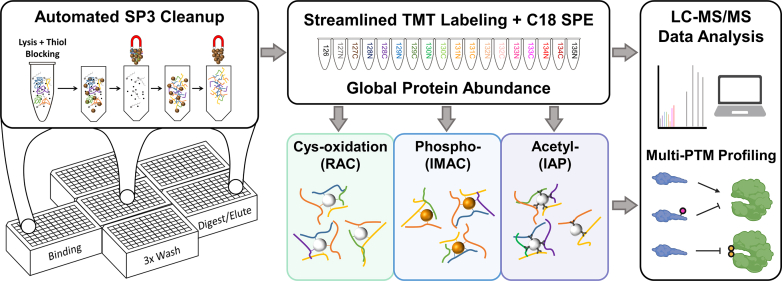
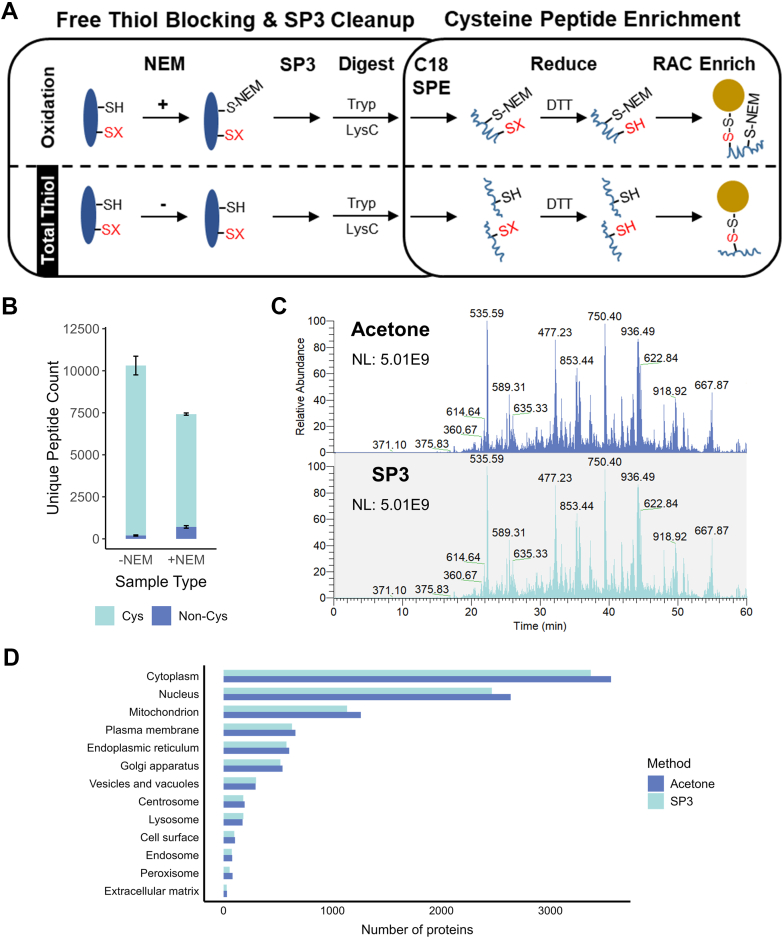
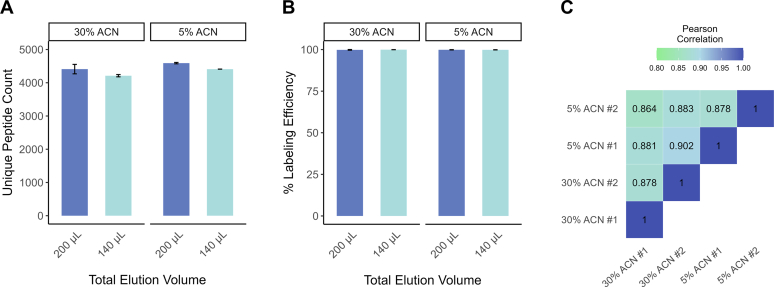
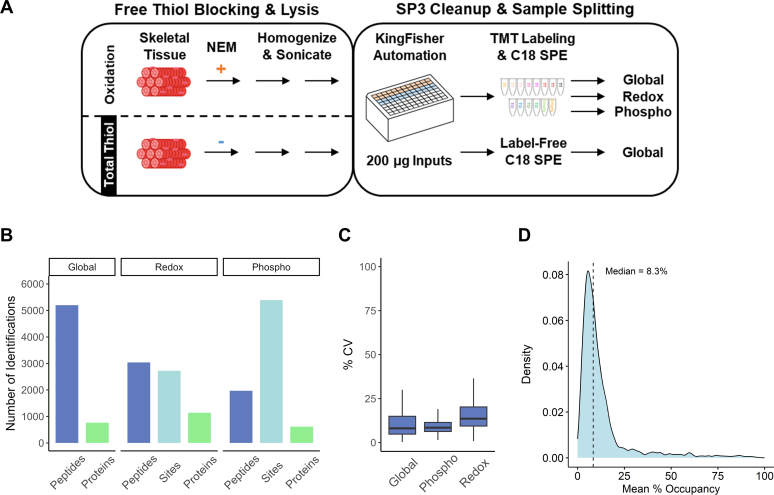
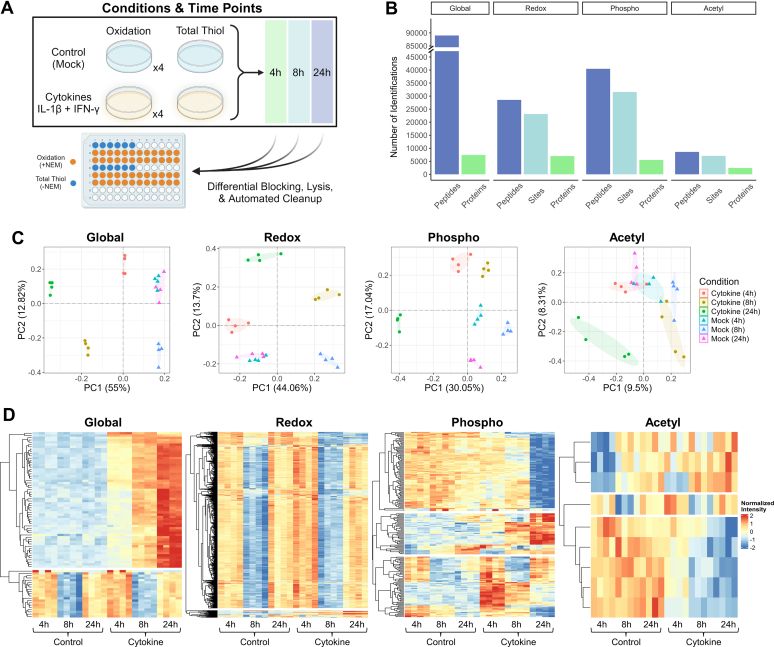
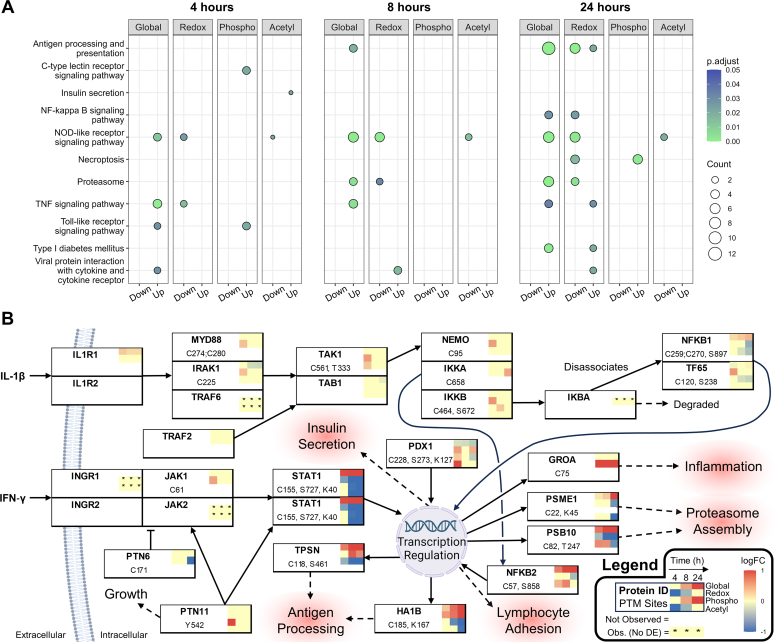
Studying regulation of protein function at a systems level necessitates an understanding of the interplay among diverse posttranslational modifications (PTMs). A variety of proteomics sample processing workflows are currently used to study specific PTMs but rarely characterize multiple types of PTMs from the same sample inputs. Method incompatibilities and laborious sample preparation steps complicate large-scale physiological investigations and can lead to variations in results. The single-pot, solid-phase-enhanced sample preparation (SP3) method for sample cleanup is compatible with different lysis buffers and amenable to automation, making it attractive for high-throughput multi-PTM profiling. Herein, we describe an integrative SP3 workflow for multiplexed quantification of protein abundance, cysteine thiol oxidation, phosphorylation, and acetylation. The broad applicability of this approach is demonstrated using cell and tissue samples, and its utility for studying interacting regulatory networks is highlighted in a time-course experiment of cytokine-treated β-cells. We observed a swift response in the global regulation of protein abundances consistent with rapid activation of JAK-STAT and NF-κB signaling pathways. Regulators of these pathways as well as proteins involved in their target processes displayed multi-PTM dynamics indicative of complex cellular response stages: acute, adaptation, and chronic (prolonged stress). PARP14, a negative regulator of JAK-STAT, had multiple colocalized PTMs that may be involved in intraprotein regulatory crosstalk. Our workflow provides a high-throughput platform that can profile multi-PTMomes from the same sample set, which is valuable in unraveling the functional roles of PTMs and their co-regulation.
在系统水平上研究蛋白质功能的调控需要了解多种翻译后修饰(PTM)之间的相互作用。目前,各种蛋白质组学样本处理工作流程用于研究特定的PTM,但很少从相同的样本输入中表征多种类型的PTM。方法不兼容性和繁琐的样本制备步骤使大规模生理研究复杂化,并可能导致结果的差异。用于样本净化的单罐、固相增强样本制备(SP3)方法与不同的裂解缓冲液兼容且适合自动化,使其对高通量多PTM分析具有吸引力。在此,我们描述了一种用于蛋白质丰度、半胱氨酸硫醇氧化、磷酸化和乙酰化多重定量的综合SP3工作流程。使用细胞和组织样本证明了该方法的广泛适用性,并在细胞因子处理的β细胞的时间进程实验中突出了其在研究相互作用调控网络方面的实用性。我们观察到蛋白质丰度的全局调控有迅速反应,这与JAK-STAT和NF-κB信号通路的快速激活一致。这些通路的调节因子以及参与其靶标过程的蛋白质表现出多PTM动态,表明复杂的细胞反应阶段:急性、适应和慢性(长期应激)。PARP14是JAK-STAT的负调节因子,有多个共定位的PTM,可能参与蛋白质内调控串扰。我们的工作流程提供了一个高通量平台,可以从同一组样本中分析多PTM组,这对于阐明PTM的功能作用及其协同调控很有价值。