Tüshaus Johanna, Eckert Stephan, Schliemann Marius, Zhou Yuxiang, Pfeiffer Pauline, Halves Christiane, Fusco Federico, Weigel Johannes, Hönikl Lisa, Butenschön Vicki, Todorova Rumyana, Rauert-Wunderlich Hilka, The Matthew, Rosenwald Andreas, Heinemann Volker, Holch Julian, Steiger Katja, Delbridge Claire, Meyer Bernhard, Weichert Wilko, Mogler Carolin, Kuhn Peer-Hendrik, Kuster Bernhard
Proteomics and Bioanalytics, School of Life Sciences, Technical University of Munich, Freising, Germany.
German Cancer Consortium (DKTK), Partner Site Munich, a Partnership between DKFZ and University Center Technical University of Munich, Munich, Germany.
EMBO J. 2025 Jan;44(1):304-329. doi: 10.1038/s44318-024-00289-w. Epub 2024 Nov 18.
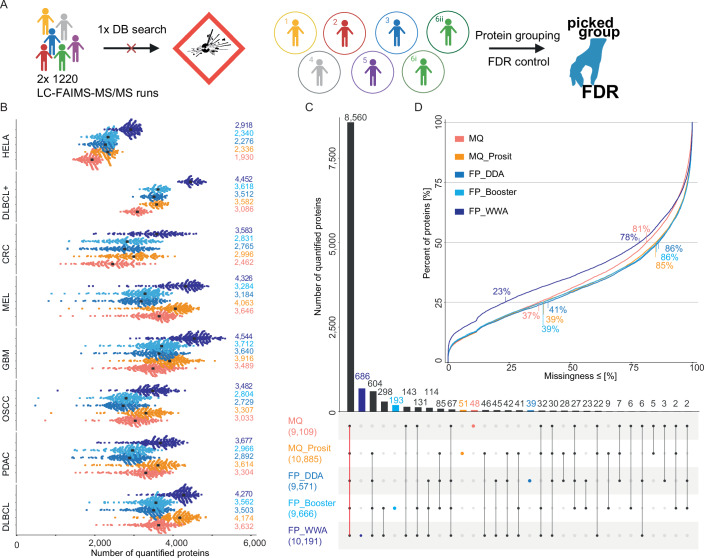
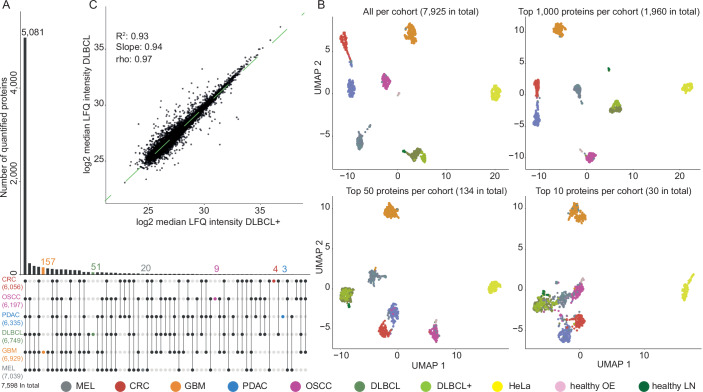
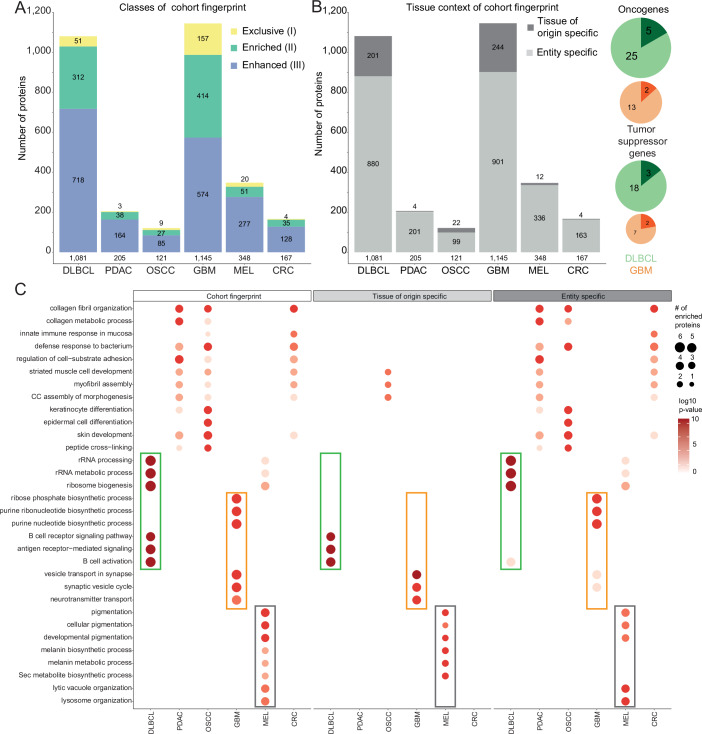
Proteome profiling of formalin-fixed paraffin-embedded (FFPE) specimens has gained traction for the analysis of cancer tissue for the discovery of molecular biomarkers. However, reports so far focused on single cancer entities, comprised relatively few cases and did not assess the long-term performance of experimental workflows. In this study, we analyze 1220 tumors from six cancer entities processed over the course of three years. Key findings include the need for a new normalization method ensuring equal and reproducible sample loading for LC-MS/MS analysis across cohorts, showing that tumors can, on average, be profiled to a depth of >4000 proteins and discovering that current software fails to process such large ion mobility-based online fractionated datasets. We report the first comprehensive pan-cancer proteome expression resource for FFPE material comprising 11,000 proteins which is of immediate utility to the scientific community, and can be explored via a web resource. It enables a range of analyses including quantitative comparisons of proteins between patients and cohorts, the discovery of protein fingerprints representing the tissue of origin or proteins enriched in certain cancer entities.
福尔马林固定石蜡包埋(FFPE)标本的蛋白质组分析在癌症组织分析以发现分子生物标志物方面已受到关注。然而,迄今为止的报告集中在单一癌症实体上,病例相对较少,且未评估实验工作流程的长期性能。在本研究中,我们分析了三年内处理的来自六个癌症实体的1220个肿瘤。主要发现包括需要一种新的归一化方法,以确保跨队列的液相色谱-串联质谱(LC-MS/MS)分析中样本加载的均匀性和可重复性;表明肿瘤平均可分析深度超过4000种蛋白质;以及发现当前软件无法处理如此大的基于离子淌度的在线分级数据集。我们报告了首个针对FFPE材料的全面泛癌蛋白质组表达资源,包含11000种蛋白质,该资源对科学界具有直接实用性,可通过网络资源进行探索。它能够进行一系列分析,包括患者和队列之间蛋白质的定量比较、发现代表组织起源的蛋白质指纹或某些癌症实体中富集的蛋白质。