Yu Fengchao, Deng Yamei, Nesvizhskii Alexey I
Department of Pathology, University of Michigan, Ann Arbor, MI, USA.
Gilbert S. Omenn Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, MI, USA.
Nat Commun. 2025 Apr 8;16(1):3329. doi: 10.1038/s41467-025-58728-z.
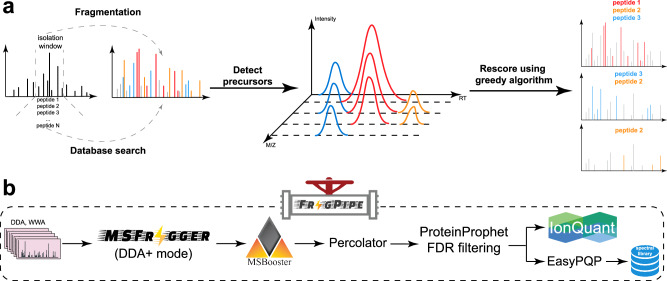
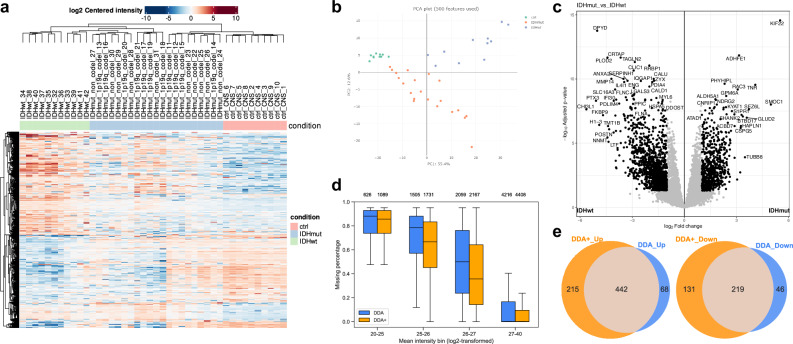
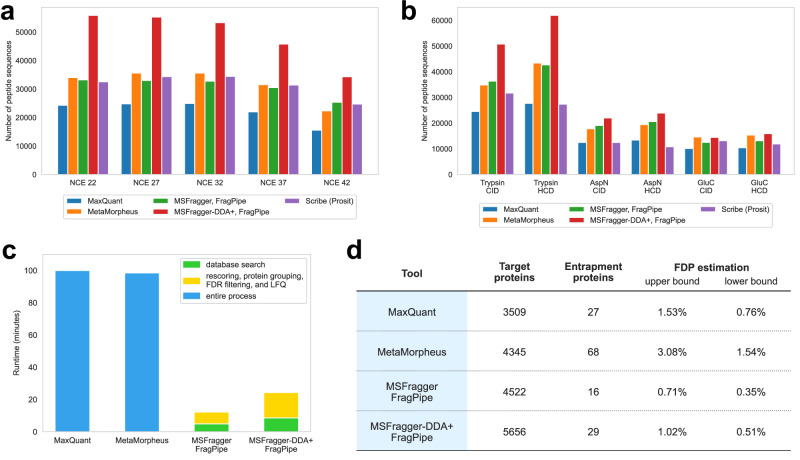
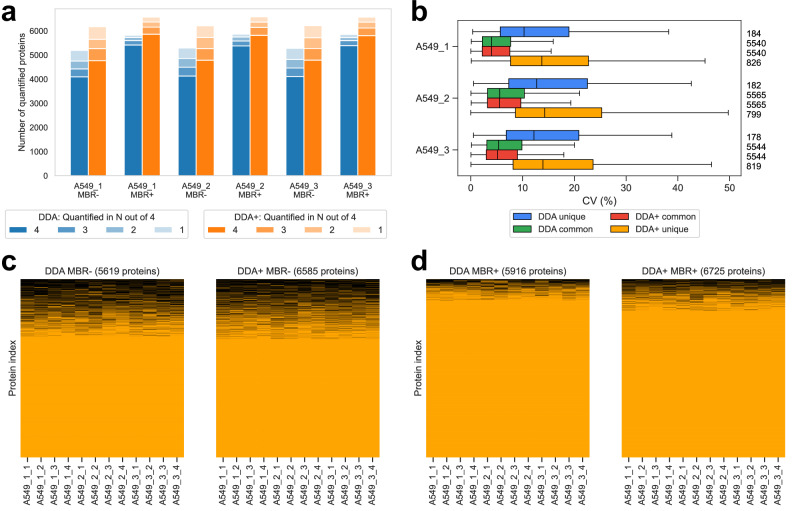
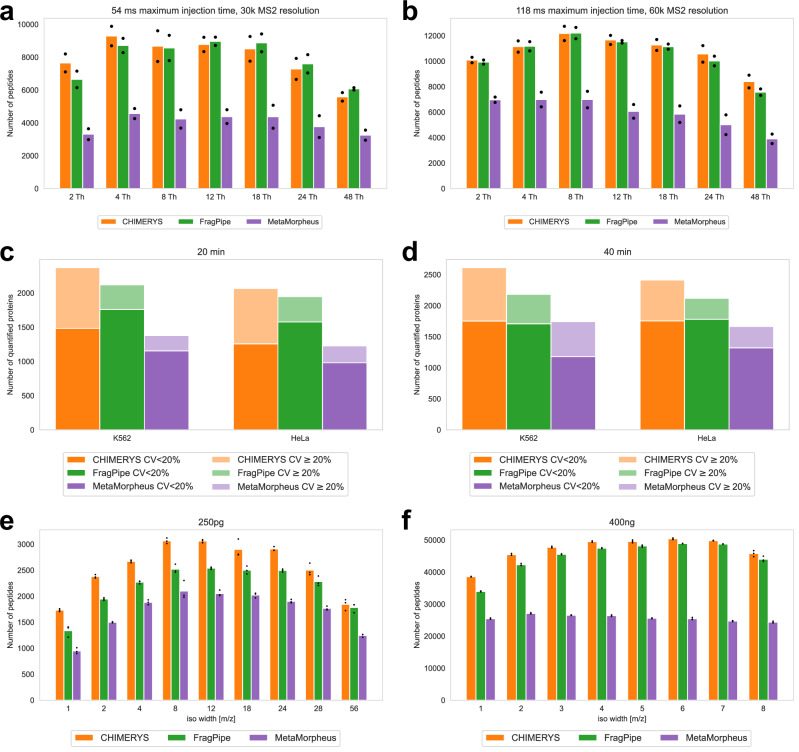
Liquid chromatography-mass spectrometry based proteomics, particularly in the bottom-up approach, relies on the digestion of proteins into peptides for subsequent separation and analysis. The most prevalent method for identifying peptides from data-dependent acquisition mass spectrometry data is database search. Traditional tools typically focus on identifying a single peptide per tandem mass spectrum, often neglecting the frequent occurrence of peptide co-fragmentations leading to chimeric spectra. Here, we introduce MSFragger-DDA+, a database search algorithm that enhances peptide identification by detecting co-fragmented peptides with high sensitivity and speed. Utilizing MSFragger's fragment ion indexing algorithm, MSFragger-DDA+ performs a comprehensive search within the full isolation window for each tandem mass spectrum, followed by robust feature detection, filtering, and rescoring procedures to refine search results. Evaluation against established tools across diverse datasets demonstrated that, integrated within the FragPipe computational platform, MSFragger-DDA+ significantly increases identification sensitivity while maintaining stringent false discovery rate control. It is also uniquely suited for wide-window acquisition data. MSFragger-DDA+ provides an efficient and accurate solution for peptide identification, enhancing the detection of low-abundance co-fragmented peptides. Coupled with the FragPipe platform, MSFragger-DDA+ enables more comprehensive and accurate analysis of proteomics data.
基于液相色谱-质谱联用的蛋白质组学,尤其是自下而上的方法,依赖于将蛋白质消化成肽段以便后续的分离和分析。从数据依赖型采集质谱数据中鉴定肽段最普遍的方法是数据库搜索。传统工具通常专注于为每个串联质谱鉴定单个肽段,常常忽略导致嵌合谱图的肽段共碎裂的频繁发生。在此,我们介绍MSFragger-DDA+,一种数据库搜索算法,它通过高灵敏度和高速度检测共碎裂肽段来增强肽段鉴定。利用MSFragger的碎片离子索引算法,MSFragger-DDA+对每个串联质谱在整个隔离窗口内进行全面搜索,随后进行强大的特征检测、过滤和重新评分程序以优化搜索结果。在不同数据集上与既定工具进行的评估表明,整合在FragPipe计算平台内,MSFragger-DDA+在保持严格的错误发现率控制的同时显著提高了鉴定灵敏度。它也特别适用于宽窗口采集数据。MSFragger-DDA+为肽段鉴定提供了一种高效且准确的解决方案,增强了对低丰度共碎裂肽段的检测。与FragPipe平台相结合,MSFragger-DDA+能够对蛋白质组学数据进行更全面和准确的分析。