Stadelmann Tobias, Heid Daniel, Jendrusch Michael, Mathony Jan, Aschenbrenner Sabine, Rosset Stéphane, Correia Bruno E, Niopek Dominik
Center for Synthetic Biology, Technical University of Darmstadt, Darmstadt 64287, Germany.
Department of Biology, Technical University of Darmstadt, Darmstadt 64287, Germany.
Nucleic Acids Res. 2024 Dec 11;52(22):e103. doi: 10.1093/nar/gkae1052.
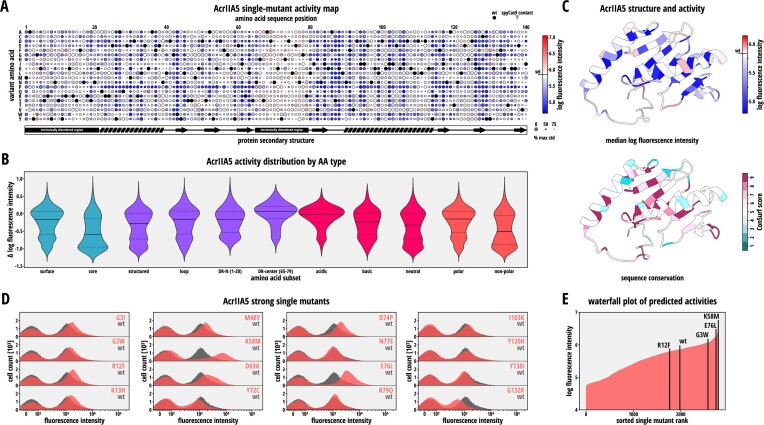
Deep mutational scanning is a powerful method for exploring the mutational fitness landscape of proteins. Its adaptation to anti-CRISPR proteins, which are natural CRISPR-Cas inhibitors and key players in the co-evolution of microbes and phages, facilitates their characterization and optimization. Here, we developed a robust anti-CRISPR deep mutational scanning pipeline in Escherichia coli that combines synthetic gene circuits based on CRISPR interference with flow cytometry coupled sequencing and mathematical modeling. Using this pipeline, we characterized comprehensive single point mutation libraries for AcrIIA4 and AcrIIA5, two potent inhibitors of CRISPR-Cas9. The resulting mutational fitness landscapes revealed considerable mutational tolerance for both Acrs, suggesting an intrinsic redundancy with respect to Cas9 inhibitory features, and - for AcrIIA5 - indicated mutations that boost Cas9 inhibition. Subsequent in vitro characterization suggested that the observed differences in inhibitory potency between mutant inhibitors were mostly due to changes in binding affinity rather than protein expression levels. Finally, to demonstrate that our pipeline can inform Acrs-based genome editing applications, we employed a selected subset of mutant inhibitors to increase CRISPR-Cas9 target specificity by modulating Cas9 activity. Taken together, our work establishes deep mutational scanning as a powerful method for anti-CRISPR protein characterization and optimization.
深度突变扫描是探索蛋白质突变适应性景观的一种强大方法。它适用于抗CRISPR蛋白,这些蛋白是天然的CRISPR-Cas抑制剂,也是微生物和噬菌体共同进化中的关键角色,有助于对抗CRISPR蛋白进行表征和优化。在这里,我们在大肠杆菌中开发了一种强大的抗CRISPR深度突变扫描流程,该流程将基于CRISPR干扰的合成基因回路与流式细胞术联合测序及数学建模相结合。利用这个流程,我们对AcrIIA4和AcrIIA5这两种强效CRISPR-Cas9抑制剂的全面单点突变文库进行了表征。由此产生的突变适应性景观揭示了这两种抗CRISPR蛋白都具有相当大的突变耐受性,表明它们在Cas9抑制特性方面存在内在冗余,并且——对于AcrIIA5——还指出了能增强Cas9抑制作用的突变。随后的体外表征表明,突变抑制剂之间观察到的抑制效力差异主要是由于结合亲和力的变化,而非蛋白质表达水平的变化。最后,为了证明我们的流程能够为基于抗CRISPR蛋白的基因组编辑应用提供信息,我们使用了选定的一组突变抑制剂,通过调节Cas9活性来提高CRISPR-Cas9的靶向特异性。综上所述,我们的工作确立了深度突变扫描作为一种用于抗CRISPR蛋白表征和优化的强大方法。