Bassi Nicolò, Xu Xiushang, Xiang Feifei, Krane Nils, Pignedoli Carlo A, Narita Akimitsu, Fasel Roman, Ruffieux Pascal
nanotech@surfaces Laboratory, Empa, Swiss Federal Laboratories for Materials Science and Technology, Dübendorf, Switzerland.
Organic and Carbon Nanomaterials Unit, Okinawa Institute of Science and Technology Graduate University, Okinawa, Japan.
Commun Chem. 2024 Nov 21;7(1):274. doi: 10.1038/s42004-024-01344-7.
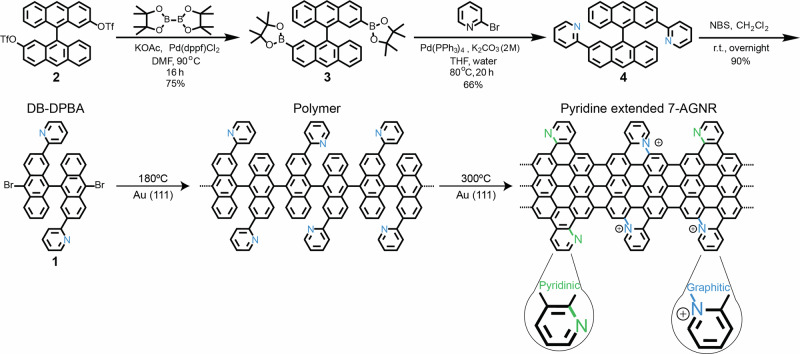
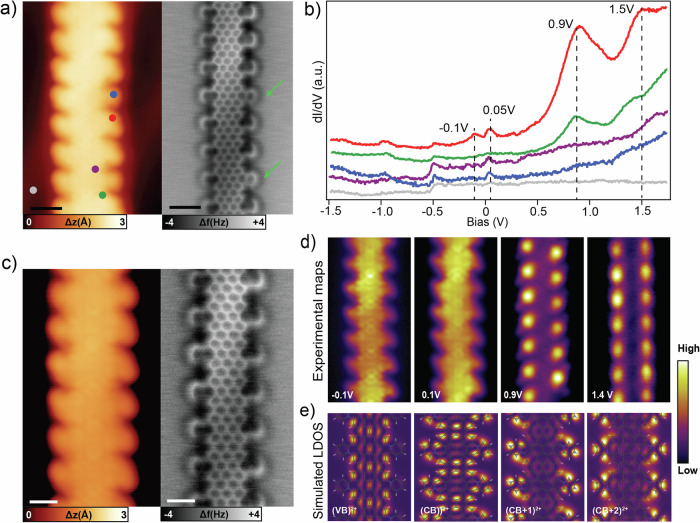
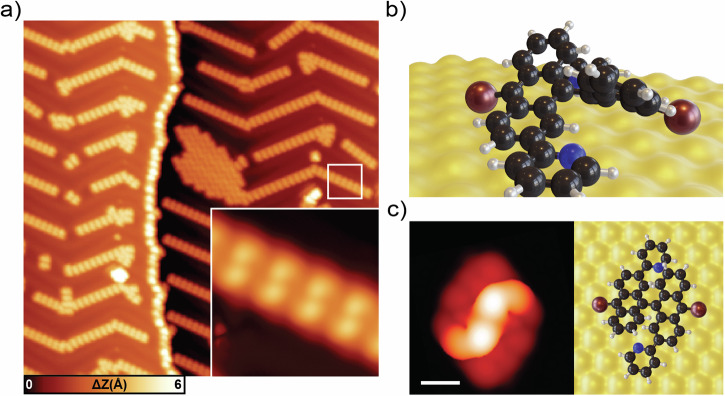
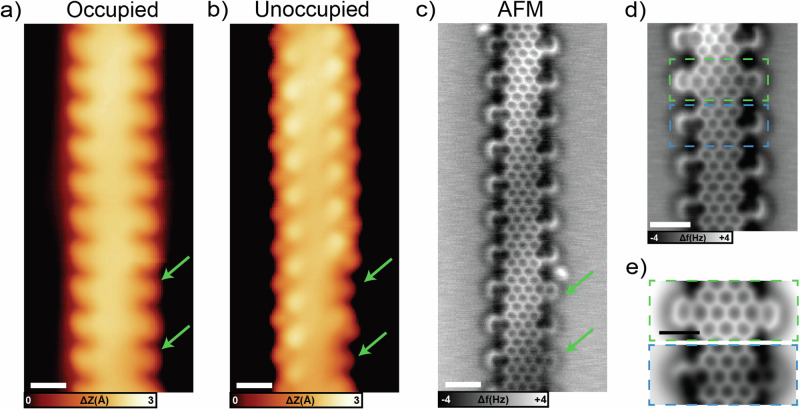
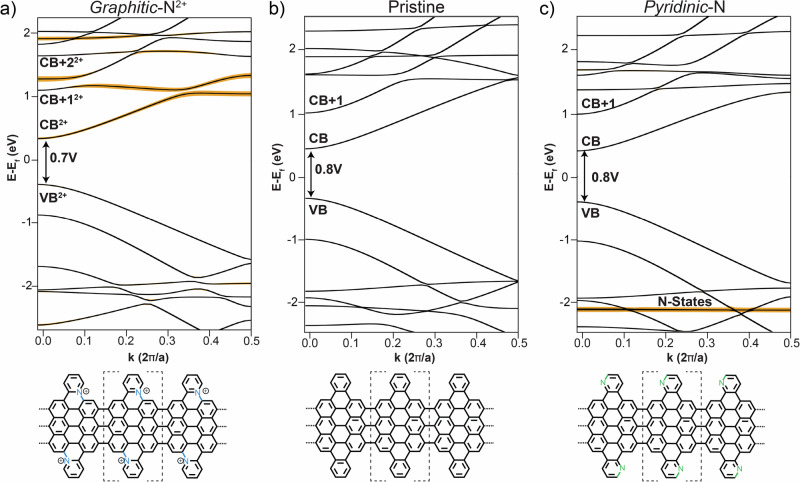
Graphene nanoribbons (GNRs), nanometer-wide strips of graphene, have garnered significant attention due to their tunable electronic and magnetic properties arising from quantum confinement. A promising approach to manipulate their electronic characteristics involves substituting carbon with heteroatoms, such as nitrogen, with different effects predicted depending on their position. In this study, we present the extension of the edges of 7-atom-wide armchair graphene nanoribbons (7-AGNRs) with pyridine rings, achieved on a Au(111) surface via on-surface synthesis. High-resolution structural characterization confirms the targeted structure, showcasing the predominant formation of carbon-nitrogen (C-N) bonds (over 90% of the units) during growth. This favored bond formation pathway is elucidated and confirmed through density functional theory (DFT) simulations. Furthermore, an analysis of the electronic properties reveals metallic behavior due to charge transfer to the Au(111) substrate accompanied by the presence of nitrogen-localized states. Our results underscore the successful formation of C-N bonds on the metal surface, providing insights for designing new GNRs that incorporate substitutional nitrogen atoms to precisely control their electronic properties.
石墨烯纳米带(GNRs)是纳米宽度的石墨烯条带,由于量子限域效应产生的可调节电子和磁性特性而备受关注。一种操纵其电子特性的有前景的方法是用杂原子(如氮)取代碳,根据其位置不同会产生不同的预测效果。在本研究中,我们展示了通过表面合成在Au(111)表面实现用吡啶环扩展7原子宽扶手椅型石墨烯纳米带(7-AGNRs)的边缘。高分辨率结构表征证实了目标结构,展示了生长过程中碳 - 氮(C - N)键的主要形成(超过90%的单元)。通过密度泛函理论(DFT)模拟阐明并证实了这种有利的键形成途径。此外,对电子性质的分析揭示了由于电荷转移到Au(111)衬底以及氮局域态的存在而导致的金属行为。我们的结果强调了在金属表面成功形成C - N键,为设计包含取代氮原子以精确控制其电子性质的新型GNRs提供了见解。