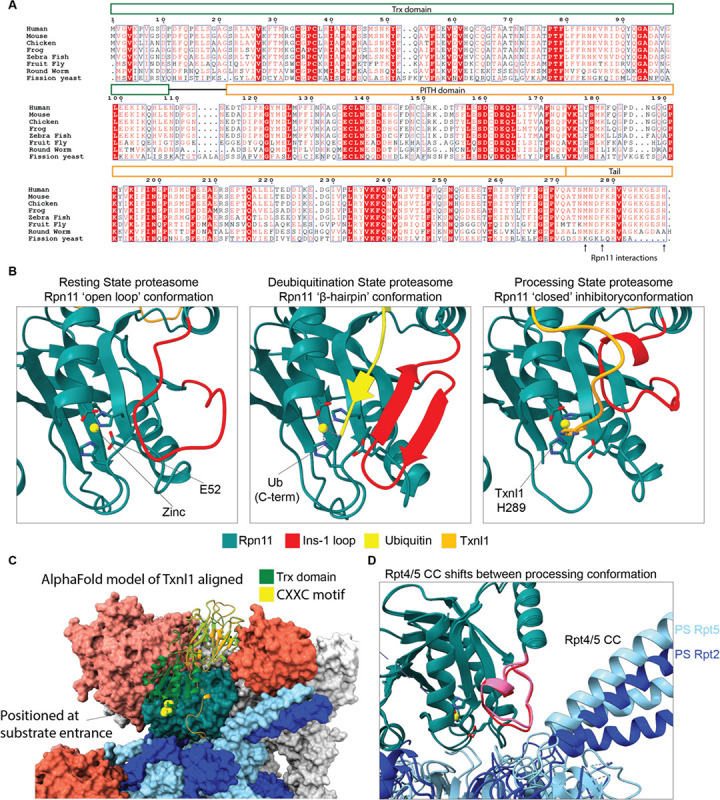
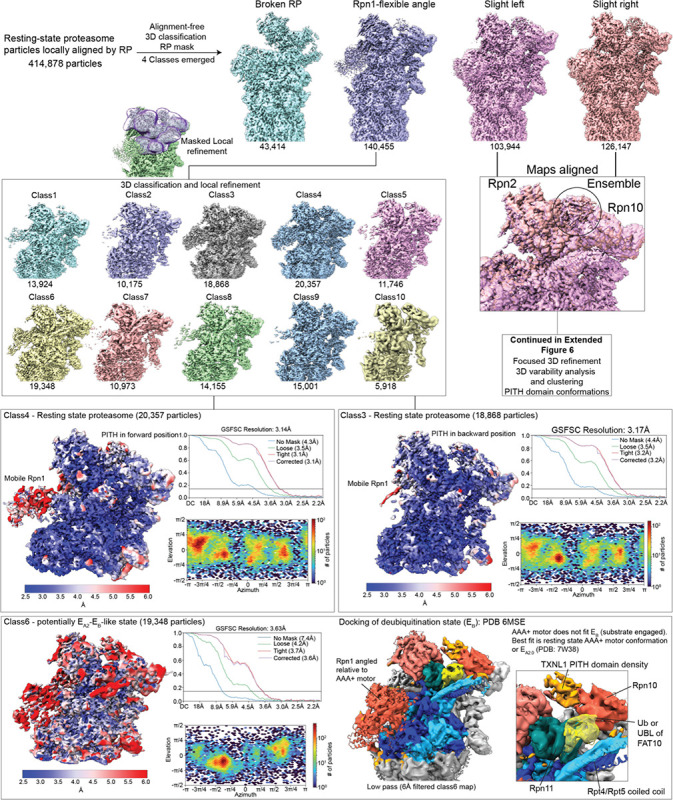
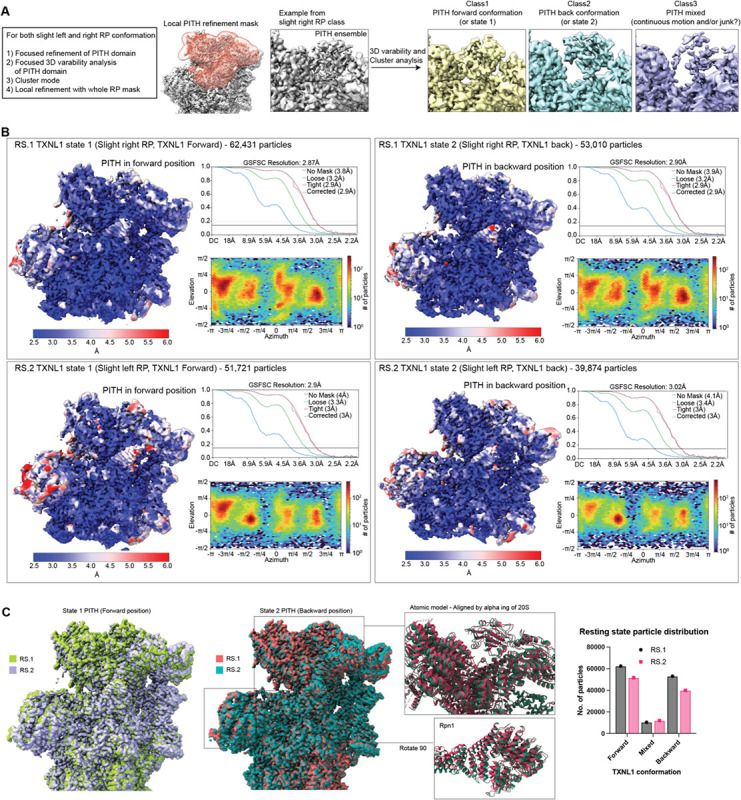
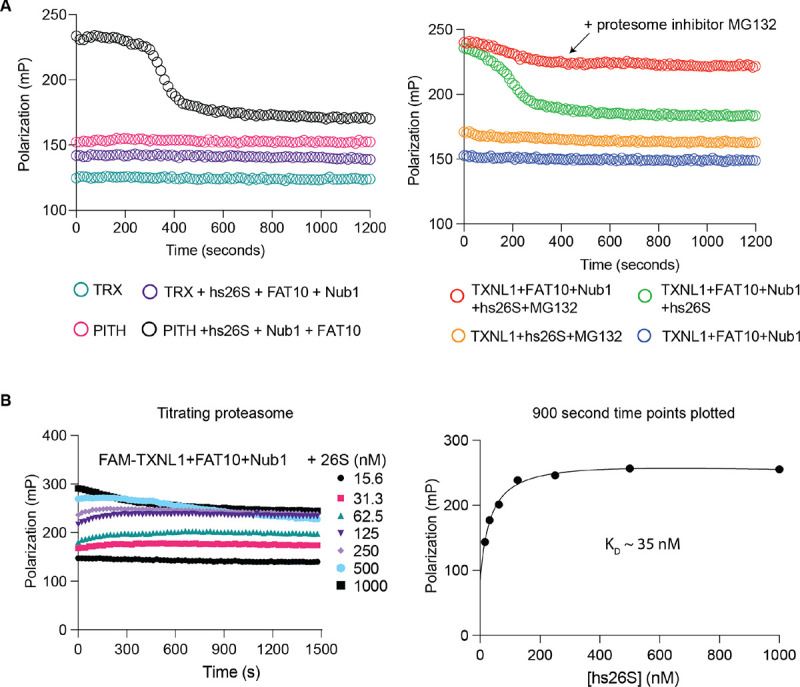
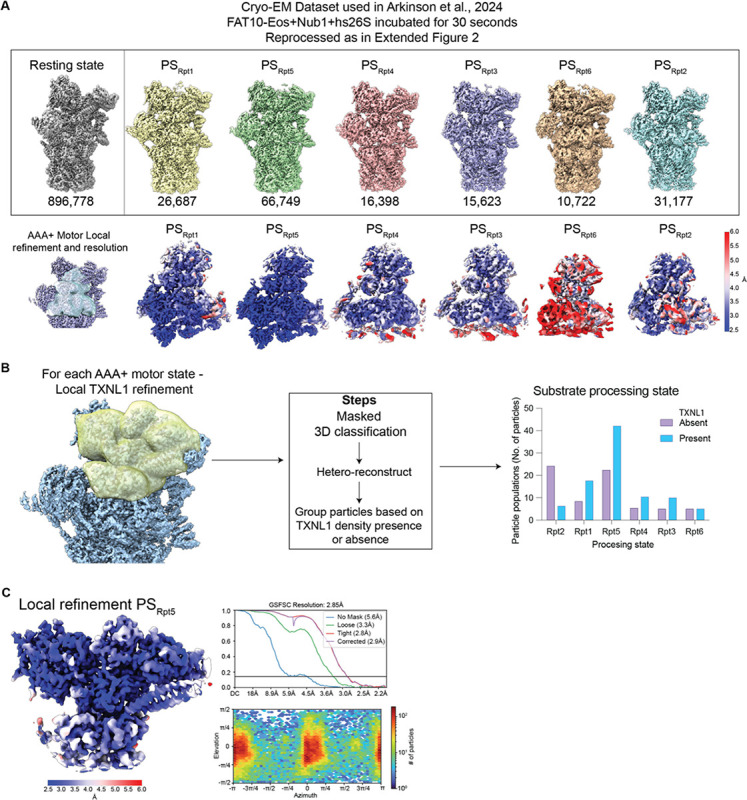
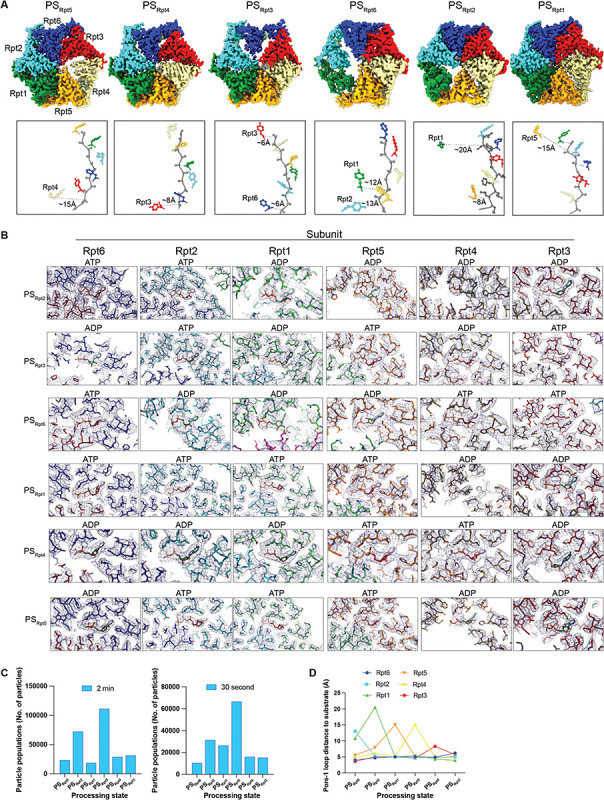
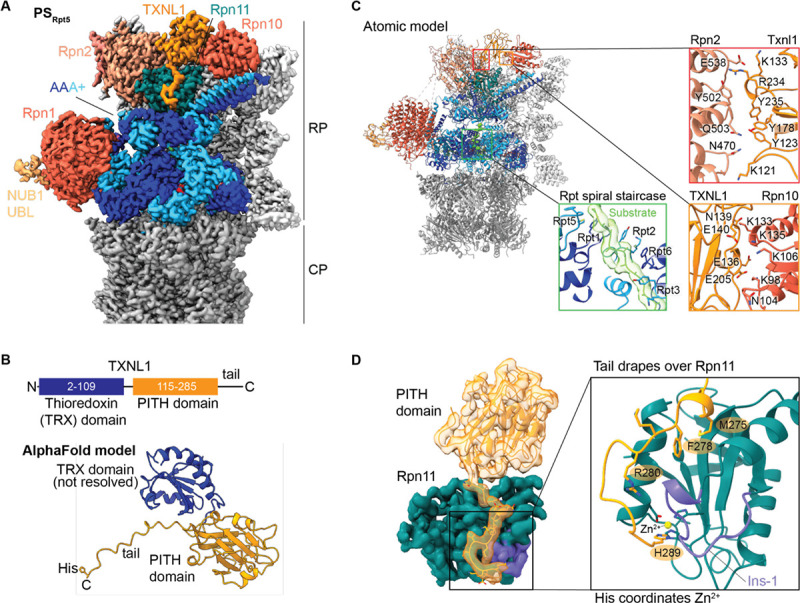
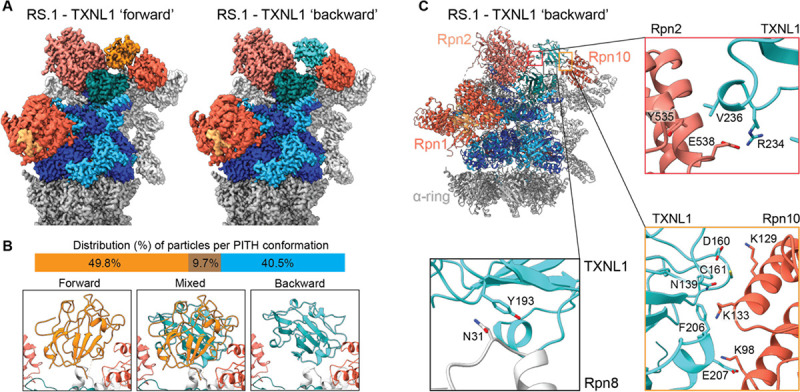
Arkinson Connor, Gee Christine L, Zhang Zeyuan, Dong Ken C, Martin Andreas
California Institute for Quantitative Biosciences, University of California at Berkeley, Berkeley, CA 94720, USA.
Department of Molecular and Cell Biology, University of California at Berkeley, Berkeley, CA 94720, USA.
bioRxiv. 2024 Nov 9:2024.11.08.622731. doi: 10.1101/2024.11.08.622731.
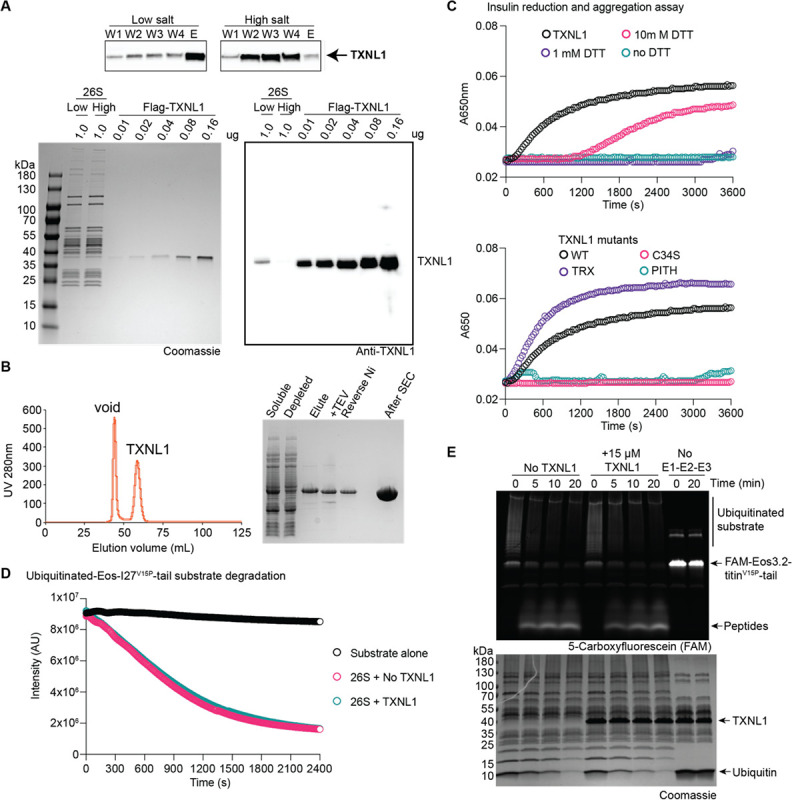
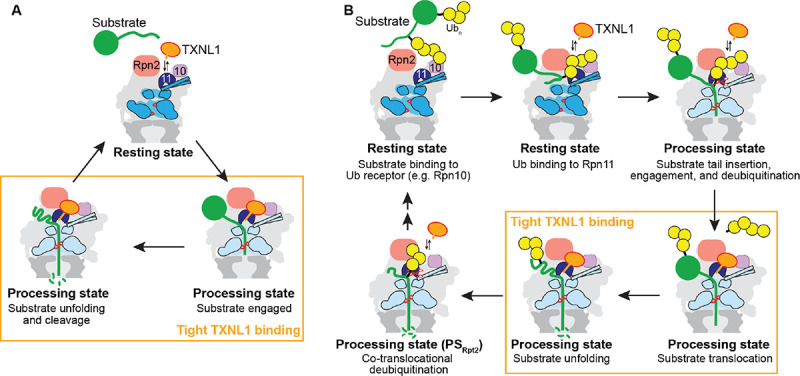
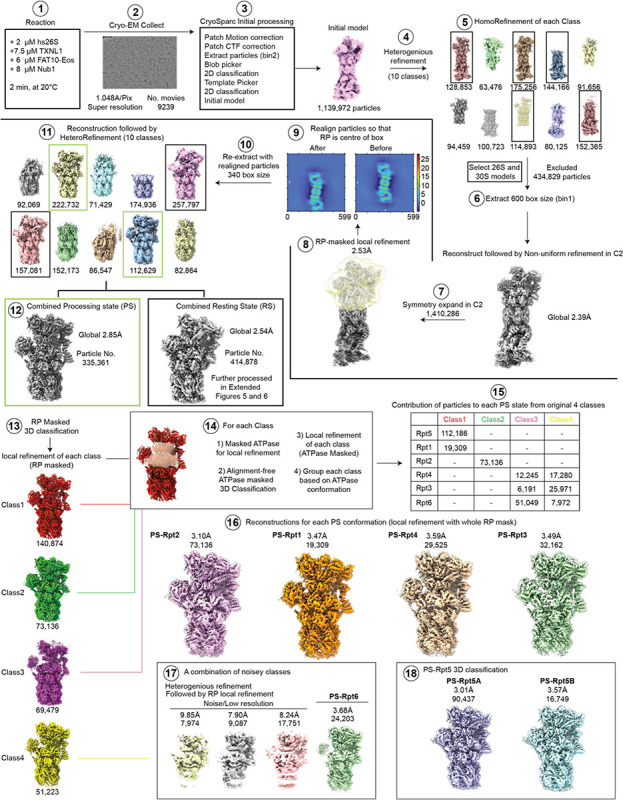
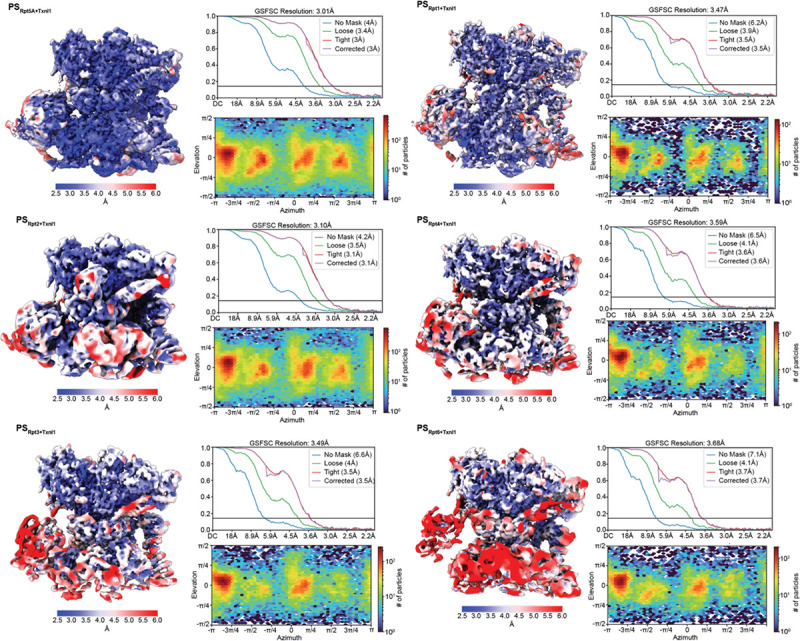
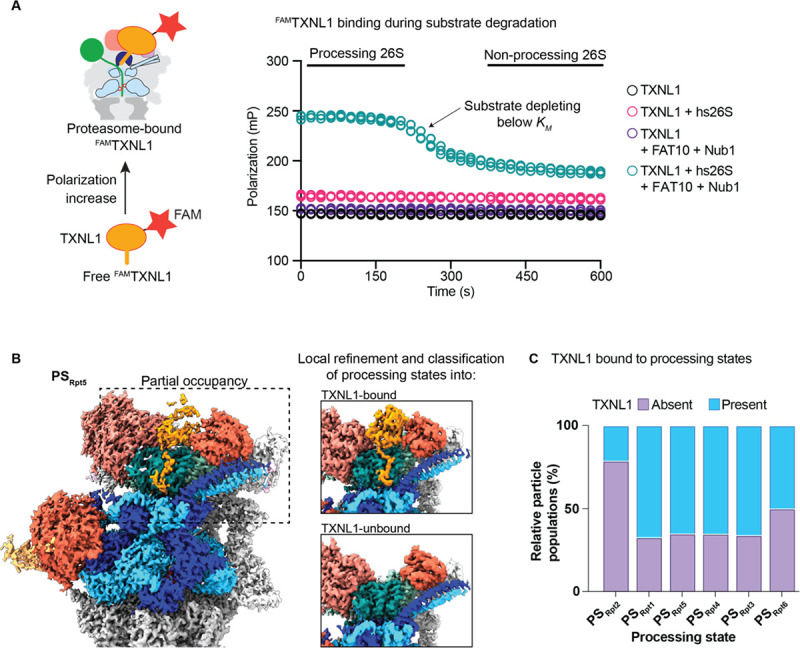
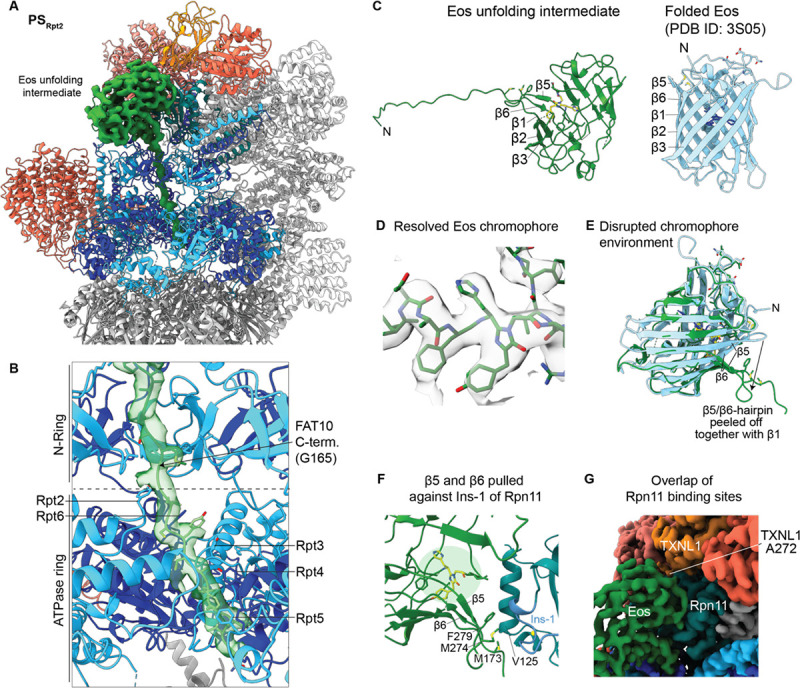
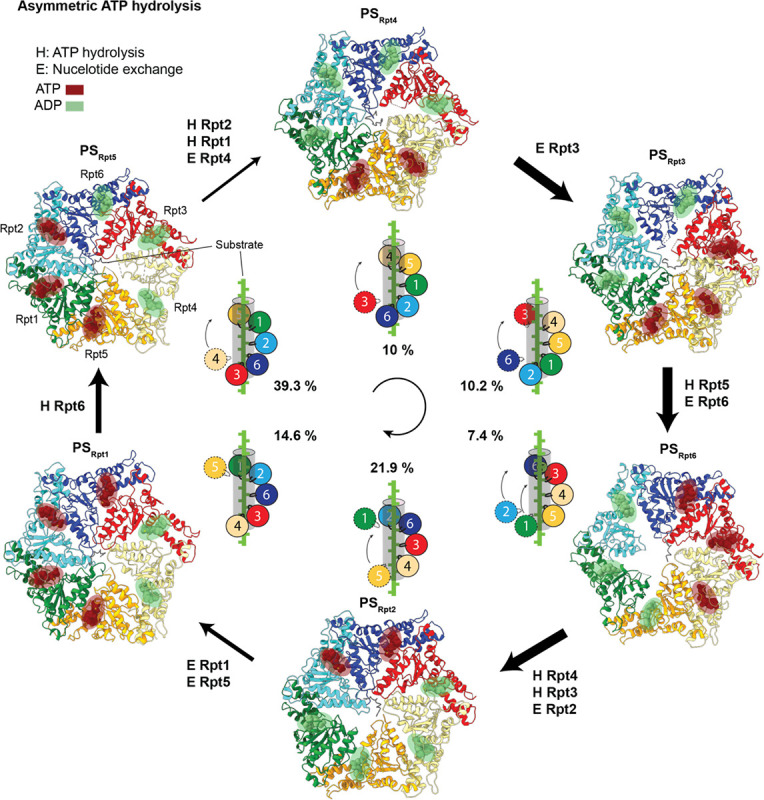
The 26S proteasome targets many cellular proteins for degradation during general homeostasis, protein quality control, and the regulation of vital processes. A broad range of proteasome-interacting cofactors thereby modulates these functions and aids in substrate degradation. Here, we solved several high-resolution structures of the redox active cofactor TXNL1 bound to the human 26S proteasome at saturating and sub-stoichiometric concentrations by time resolved cryo-EM. We identified distinct binding modes of TXNL1 that depend on the proteasome conformational and ATPase motor states. Together with biophysical and biochemical experiments, our structural studies reveal that the resting-state proteasome prior to substrate engagement with the ATPase motor binds TXNL1 with low affinity and in variable positions on top of the Rpn11 deubiquitinase. In contrast, the actively degrading proteasome shows additional interactions leading to high-affinity TXNL1 binding, whereby TXNL1's C-terminal tail covers the catalytic groove of the Rpn11 deubiquitinase and coordinates the active-site Zn. Furthermore, these cryo-EM structures of the degrading proteasome capture the ATPase hexamer in all registers of spiral-staircase arrangements and thus visualize the complete ATP-hydrolysis cycle of the AAA+ motor, indicating temporally asymmetric hydrolysis and conformational changes in bursts during mechanical substrate unfolding and translocation. Remarkably, we catch the proteasome in the act of unfolding the beta-barrel mEos3.2 substrate while the ATPase hexamer is in a particular spiral staircase register. Our findings challenge current models for protein translocation through hexameric AAA+ motors and reveal how the proteasome uses its distinct but broad range of conformational states to coordinate cofactor binding and substrate processing.
在一般的体内平衡、蛋白质质量控制和重要过程的调节过程中,26S蛋白酶体靶向许多细胞蛋白进行降解。因此,多种与蛋白酶体相互作用的辅因子调节这些功能并协助底物降解。在这里,我们通过时间分辨冷冻电镜解析了氧化还原活性辅因子TXNL1在饱和和亚化学计量浓度下与人26S蛋白酶体结合的几个高分辨率结构。我们确定了TXNL1不同的结合模式,这些模式取决于蛋白酶体的构象和ATP酶马达状态。结合生物物理和生化实验,我们的结构研究表明,在底物与ATP酶马达结合之前的静止状态蛋白酶体以低亲和力结合TXNL1,且结合位置在Rpn11去泛素酶顶部可变。相反,处于活跃降解状态的蛋白酶体显示出额外的相互作用,导致与TXNL1高亲和力结合,其中TXNL1的C末端尾巴覆盖Rpn11去泛素酶的催化凹槽并配位活性位点的锌。此外,这些降解状态蛋白酶体的冷冻电镜结构捕捉到了处于螺旋楼梯排列所有寄存器状态的ATP酶六聚体,从而可视化了AAA+马达完整的ATP水解循环,表明在机械性底物解折叠和转运过程中,ATP水解和构象变化在爆发时有时间上的不对称性。值得注意的是,当ATP酶六聚体处于特定的螺旋楼梯寄存器状态时,我们捕捉到蛋白酶体正在解折叠β桶状mEos3.2底物。我们的发现挑战了当前关于蛋白质通过六聚体AAA+马达转运的模型,并揭示了蛋白酶体如何利用其独特但广泛的构象状态来协调辅因子结合和底物加工。