Shah Kinnary, Guo Boyi, Hicks Stephanie C
Department of Biostatistics, Johns Hopkins Bloomberg School of Public Health, Baltimore, MD, USA.
Department of Biomedical Engineering, Johns Hopkins School of Medicine, Baltimore, MD, USA.
bioRxiv. 2024 Nov 8:2024.11.04.621867. doi: 10.1101/2024.11.04.621867.
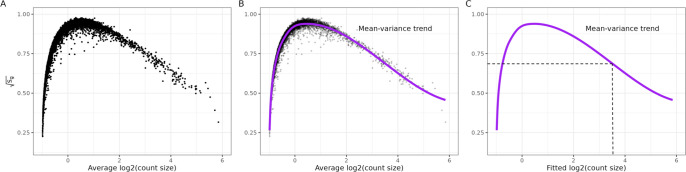
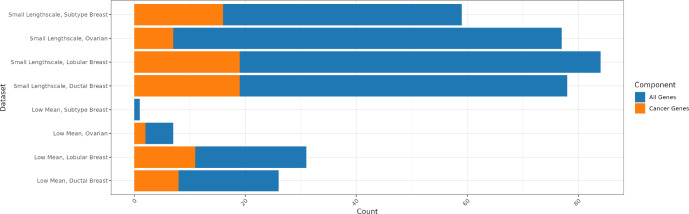
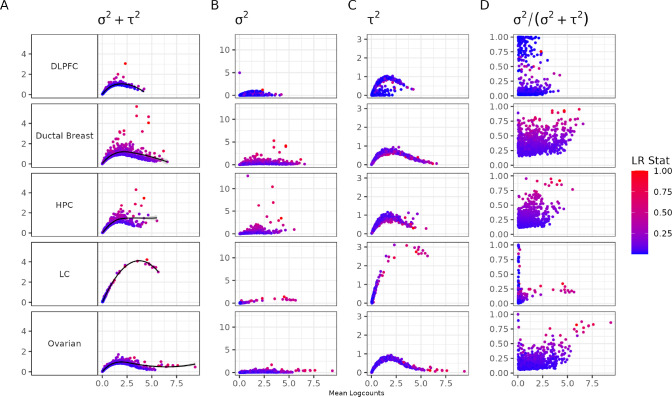
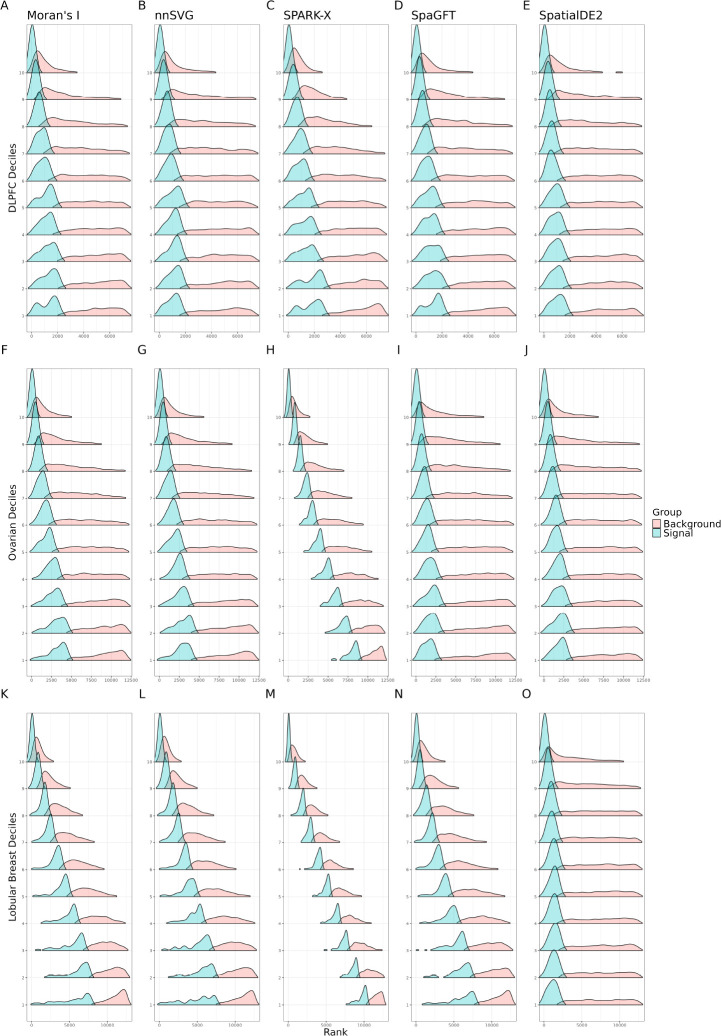
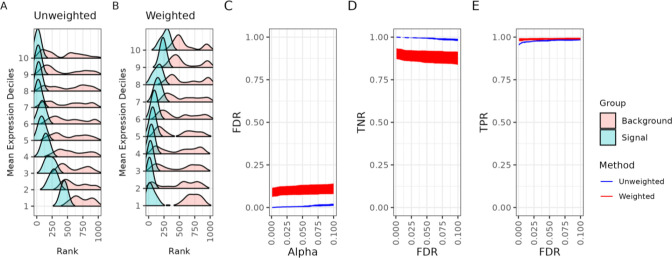
An important task in the analysis of spatially resolved transcriptomics data is to identify spatially variable genes (SVGs), or genes that vary in a 2D space. Current approaches rank SVGs based on either -values or an effect size, such as the proportion of spatial variance. However, previous work in the analysis of RNA-sequencing identified a technical bias, referred to as the "mean-variance relationship", where highly expressed genes are more likely to have a higher variance. Here, we demonstrate the mean-variance relationship in spatial transcriptomics data. Furthermore, we propose , a statistical framework using Empirical Bayes techniques to remove this bias, leading to more accurate prioritization of SVGs. We demonstrate the performance of in both simulated and real spatial transcriptomics data. A software implementation of our method is available at https://bioconductor.org/packages/spoon.
在空间分辨转录组学数据分析中的一项重要任务是识别空间可变基因(SVG),即在二维空间中变化的基因。当前的方法基于P值或效应大小(如空间方差比例)对SVG进行排名。然而,先前在RNA测序分析中的工作发现了一种技术偏差,称为“均值 - 方差关系”,即高表达基因更有可能具有更高的方差。在这里,我们展示了空间转录组学数据中的均值 - 方差关系。此外,我们提出了Spoon,这是一个使用经验贝叶斯技术来消除这种偏差的统计框架,从而更准确地对SVG进行优先级排序。我们在模拟和真实的空间转录组学数据中展示了Spoon的性能。我们方法的软件实现可在https://bioconductor.org/packages/spoon获取。