Lindahl Sarah E, Metzger Erin M, Chen Chun-Hsing, Pink Maren, Zaleski Jeffrey M
Department of Chemistry, Indiana University Bloomington IN 47405 USA
Molecular Structure Center, Indiana University Bloomington IN 47405 USA.
Chem Sci. 2024 Nov 12;16(1):255-279. doi: 10.1039/d4sc05396f. eCollection 2024 Dec 18.
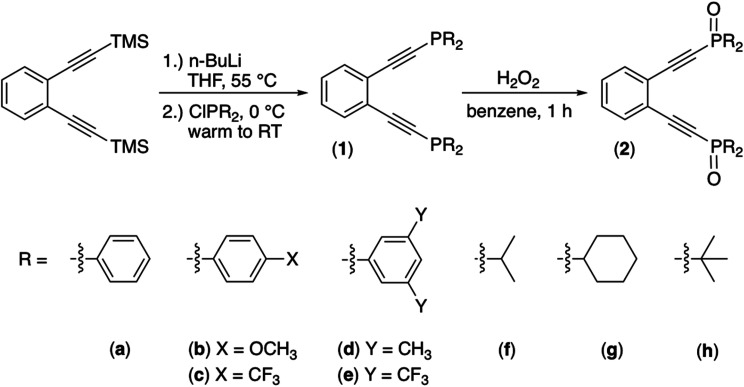
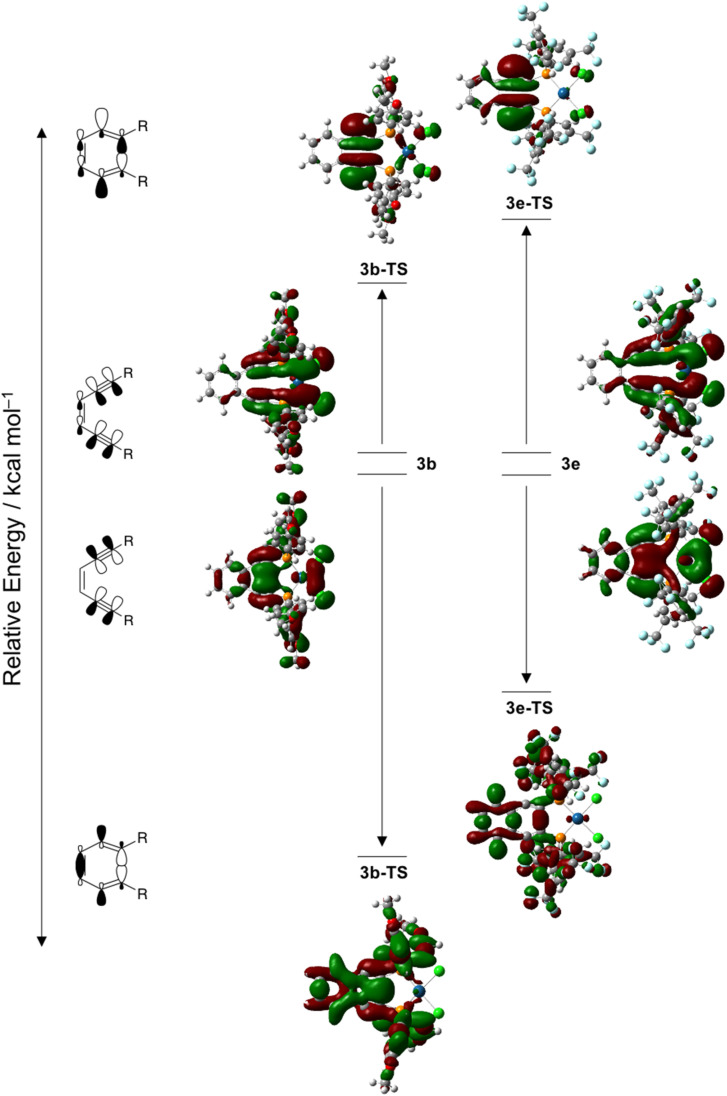
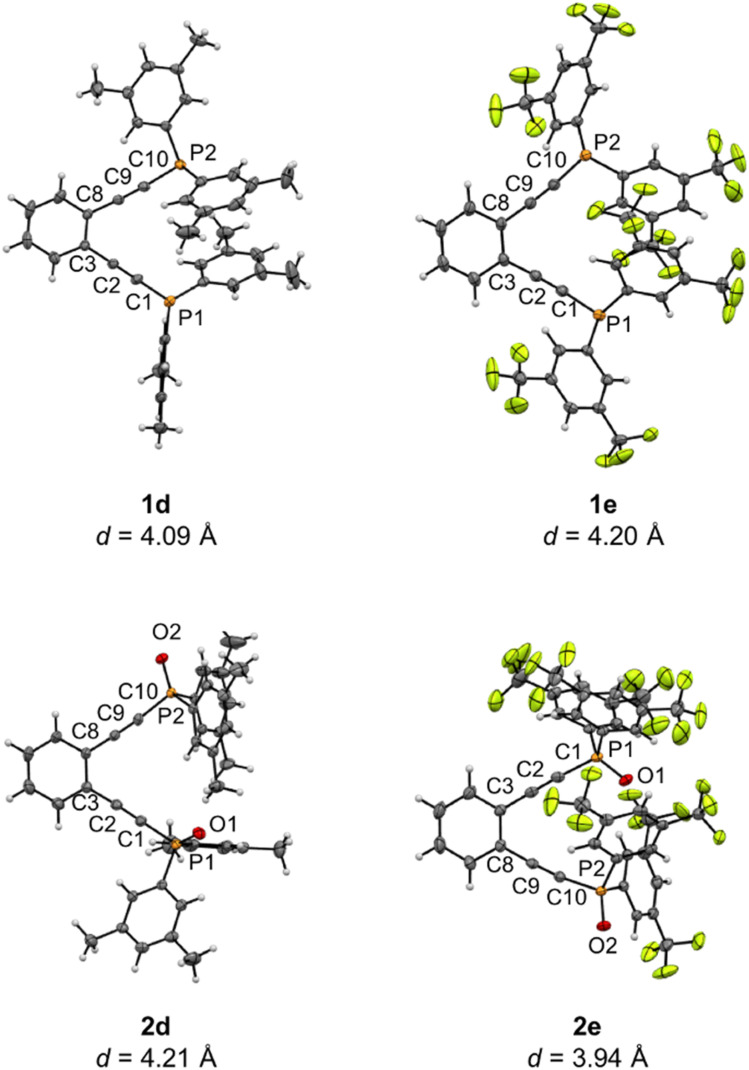
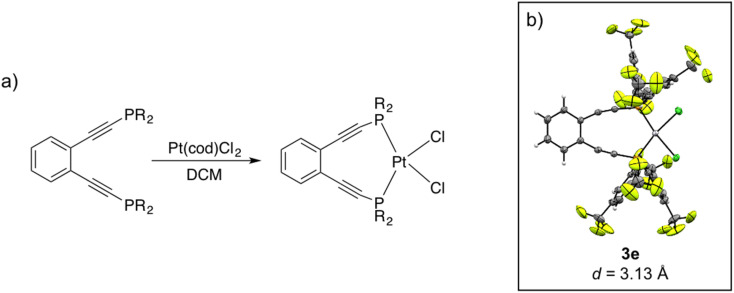
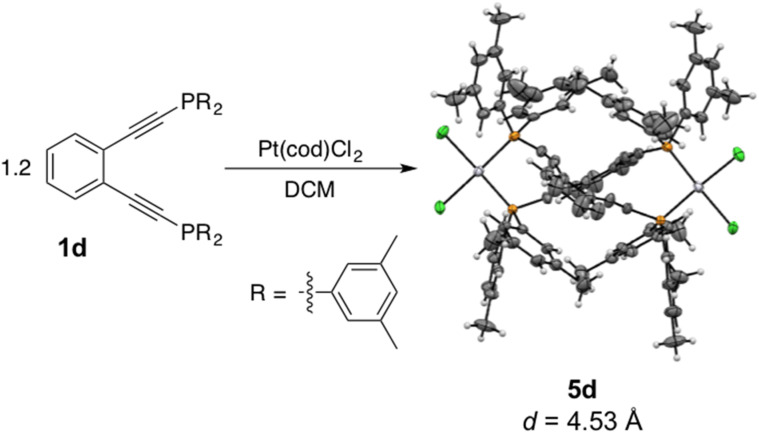
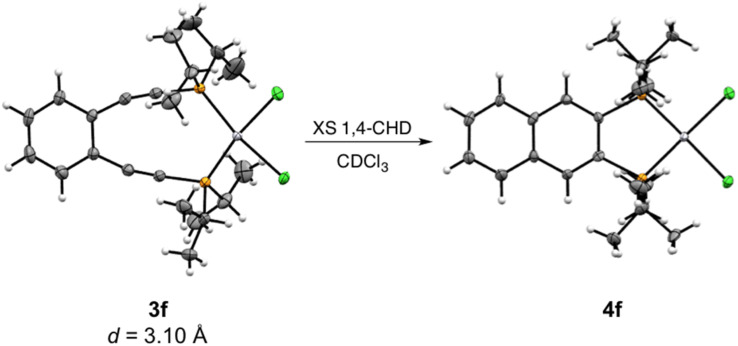
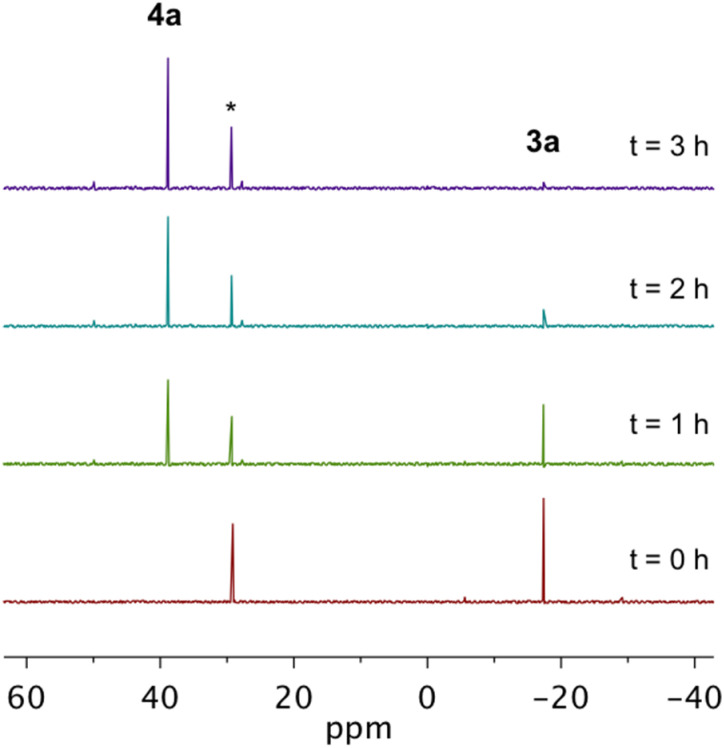
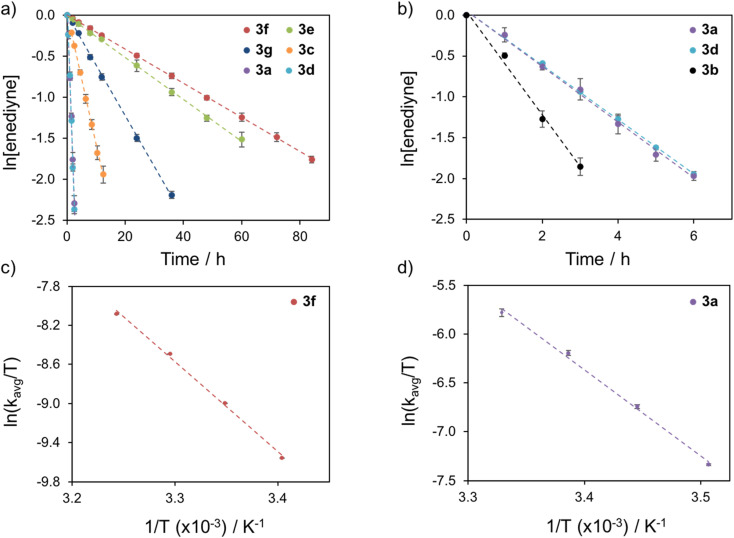
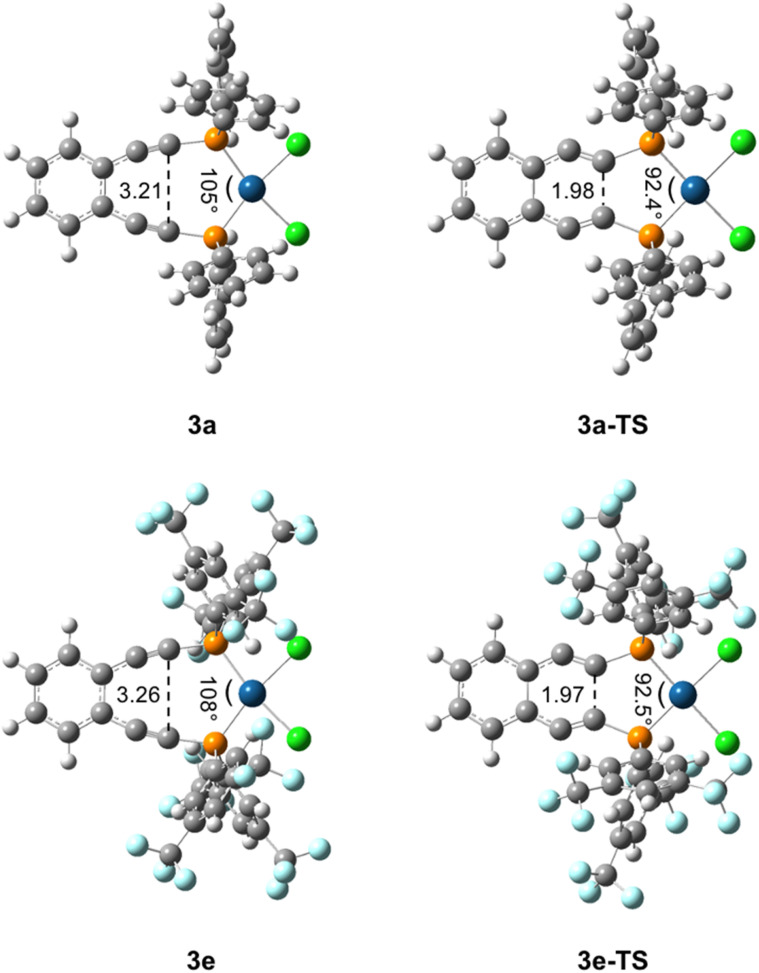
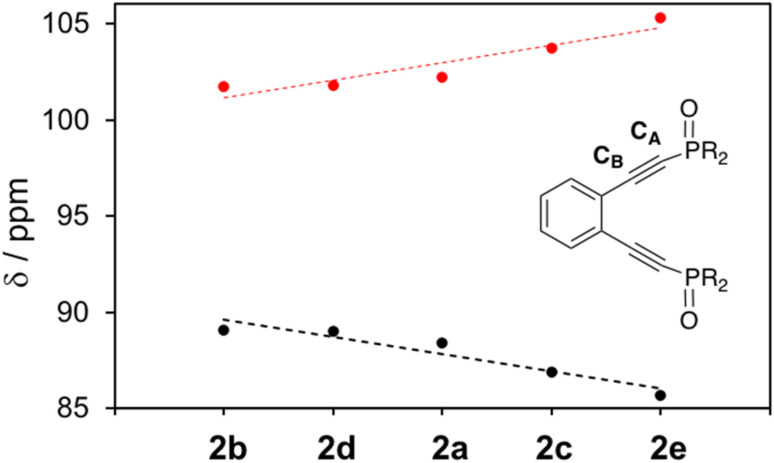
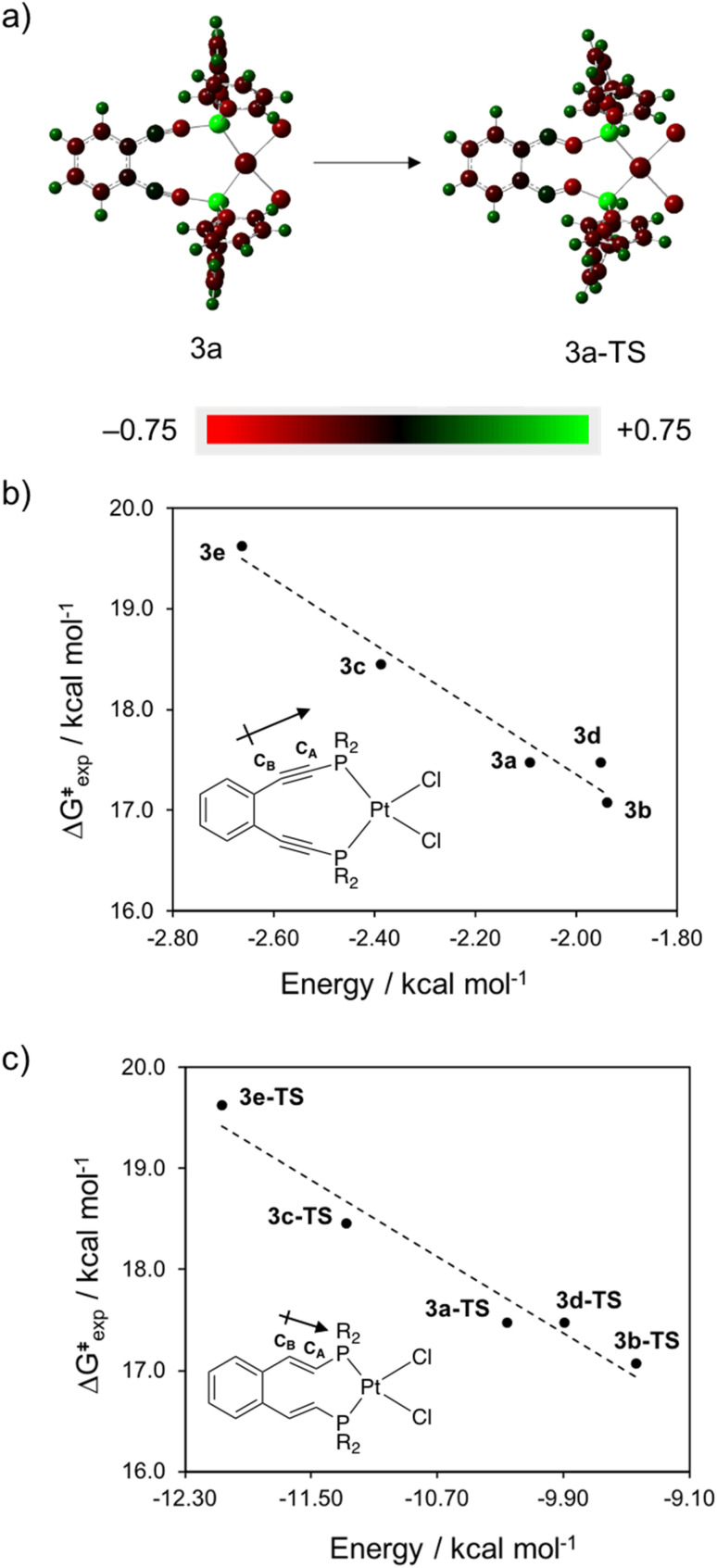
Using a diverse array of thermally robust phosphine enediyne ligands (dxpeb, X = Ph, Ph-OCH, Ph-CF, Ph- CH, Ph- CF, Pr, Cy, and Bu) a novel suite of cisplatin-like Pt(ii) metalloenediynes (3, Pt(dxpeb)Cl) has been synthesized and represents unique electronic perturbations on thermal Bergman cyclization kinetics. Complexes 3e (Ph- CF) and 3f (Pr) are the first of this structure type to be crystallographically characterized with inter alkyne termini distances (3e: 3.13 Å; 3f: 3.10 Å) at the lower end of the widely accepted critical distance range within which enediynes should demonstrate spontaneous ambient temperature cyclization. Despite different electronic profiles, these metalloenediynes adopt a rigid, uniform structure suggesting complexes of the form Pt(dxpeb)Cl have orthogonalized geometric and electronic contributions to thermal Bergman cyclization. Kinetic activation parameters determined using P NMR spectroscopy highlight the dramatic reactivity and thermal tunability of these complexes. At room temperature, the half-life ( ) of cyclization spans a range of ∼35 hours and for the aryl phosphine derivatives, cycloaromatization rates are 10-30 times faster for complexes with electron donating substituents (3b: Ph-OCH; 3d: Ph- CH) compared to those with electron withdrawing substituents (3c: Ph-CF; 3e: Ph- CF). Computational interrogation of the aryl phosphine metalloenediynes 3a-3e reveals that the origin of this precise electronic control derives from electronic withdrawing group-mediated alkyne carbon polarization that amplifies coulombic repulsion increasing the cyclization barrier height. Additionally, mixing between the in-plane π-orbitals and the phosphine aryl ring system is pronounced for complexes with electron donating substituents which stabilizes the developing C-C bond and lowers the activation barrier. This π-orbital mixing is negligible however, for complexes with electron withdrawing substituents due to an energetic mismatch of the orbital systems. Overall, this work demonstrates that for geometrically rigid frameworks, even remote enediyne functionalization can have pronounced effects on activation barrier.
通过使用一系列热稳定性良好的膦烯二炔配体(dxpeb,X = Ph、Ph - OCH、Ph - CF、Ph - CH、Ph - CF、Pr、Cy和Bu),合成了一组新型的类顺铂Pt(ii)金属烯二炔(3,Pt(dxpeb)Cl),它们在热伯格曼环化动力学上表现出独特的电子扰动。配合物3e(Ph - CF)和3f(Pr)是该结构类型中首批通过晶体学表征的,其炔端间距(3e:3.13 Å;3f:3.10 Å)处于广泛接受的临界距离范围下限,在此范围内烯二炔应表现出自发的室温环化。尽管电子性质不同,但这些金属烯二炔采用刚性、统一的结构,表明Pt(dxpeb)Cl形式的配合物对热伯格曼环化具有正交的几何和电子贡献。使用³¹P NMR光谱测定的动力学活化参数突出了这些配合物显著的反应活性和热可调性。在室温下,环化的半衰期(t₁/₂)跨度约为35小时,对于芳基膦衍生物,与具有吸电子取代基的配合物(3c:Ph - CF;3e:Ph - CF)相比,具有供电子取代基的配合物(3b:Ph - OCH;3d:Ph - CH)的环芳构化速率快10 - 30倍。对芳基膦金属烯二炔3a - 3e的计算研究表明,这种精确电子控制的起源源于吸电子基团介导的炔烃碳极化,它放大了库仑排斥力,增加了环化势垒高度。此外,对于具有供电子取代基的配合物,面内π轨道与膦芳环系统之间的混合很明显,这稳定了正在形成的C - C键并降低了活化势垒。然而,对于具有吸电子取代基的配合物,由于轨道系统的能量不匹配,这种π轨道混合可以忽略不计。总体而言,这项工作表明,对于几何刚性框架,即使是远程烯二炔官能化也会对活化势垒产生显著影响。