Alruhaimi Reem S, Mahmoud Ayman M, Alnasser Sulaiman M, Alotaibi Mohammed F, Elbagory Ibrahim, El-Bassuony Ashraf A, Lamsabhi Al Mokhtar, Kamel Emadeldin M
Department of Biology, College of Science, Princess Nourah bint Abdulrahman University, Riyadh 11671, Saudi Arabia.
Department of Life Sciences, Faculty of Science and Engineering, Manchester Metropolitan University, Manchester M1 5GD, U.K.
ACS Omega. 2024 Nov 13;9(47):47167-47179. doi: 10.1021/acsomega.4c07624. eCollection 2024 Nov 26.
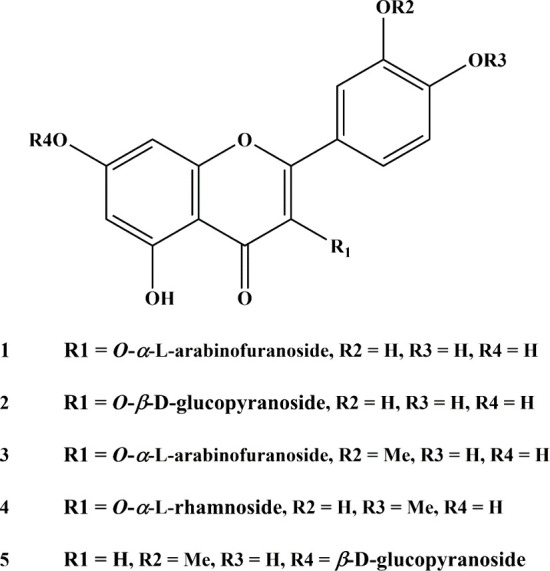
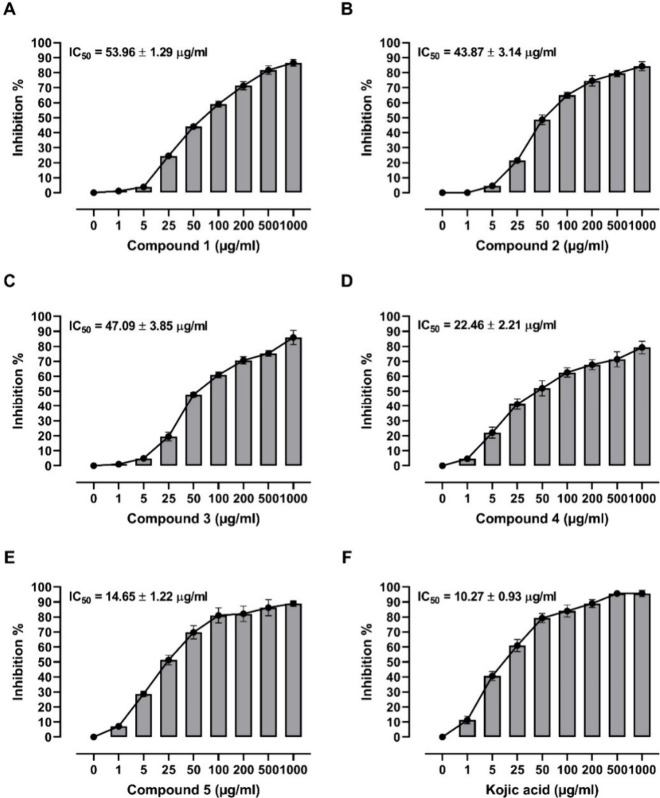
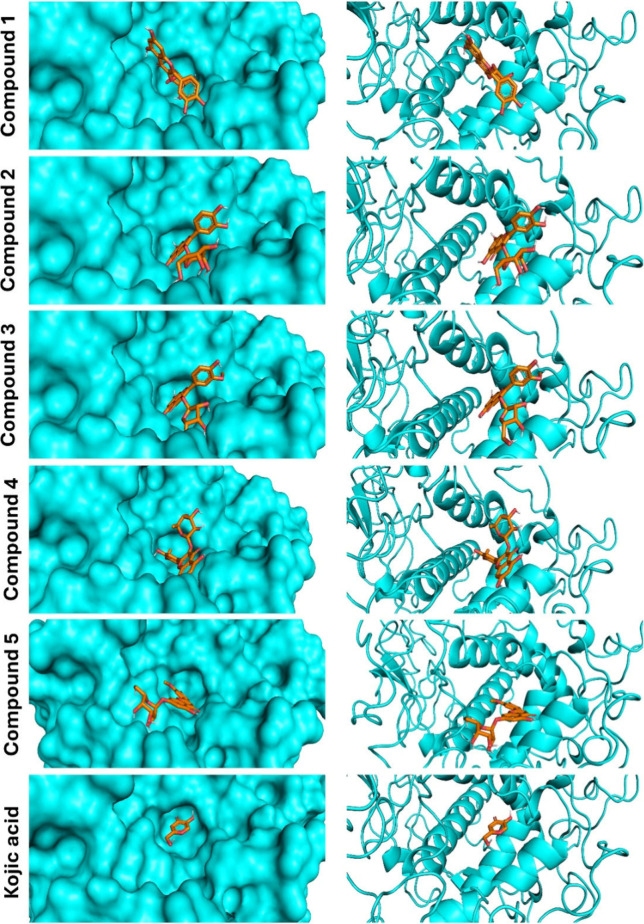
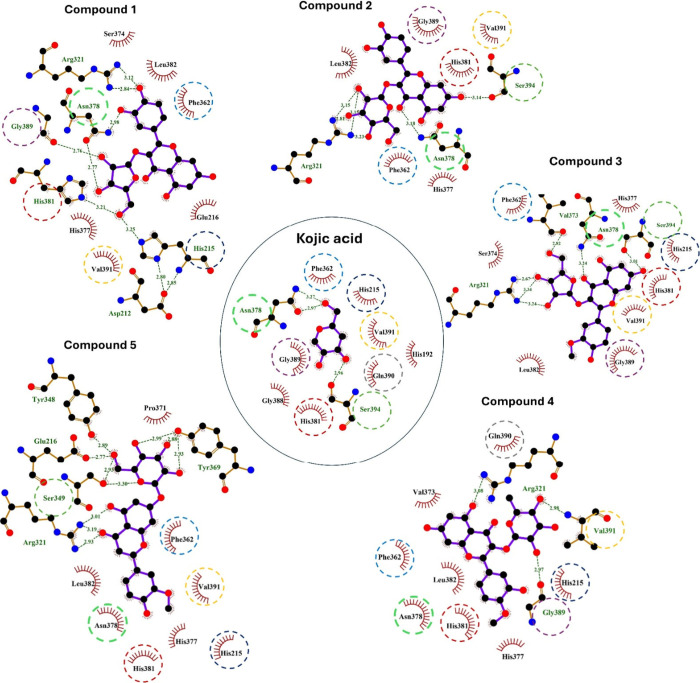
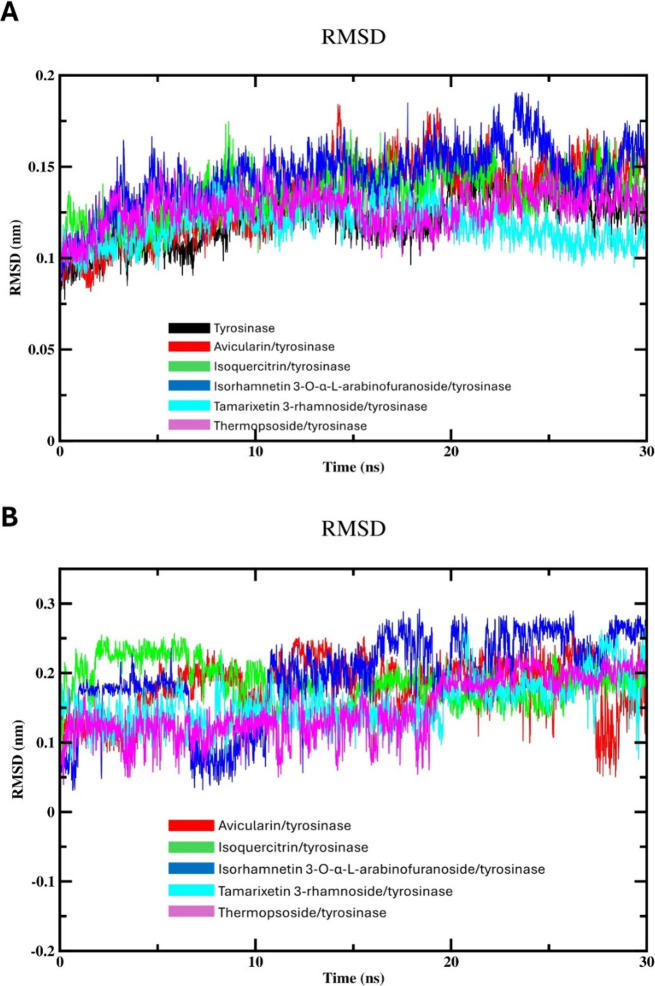
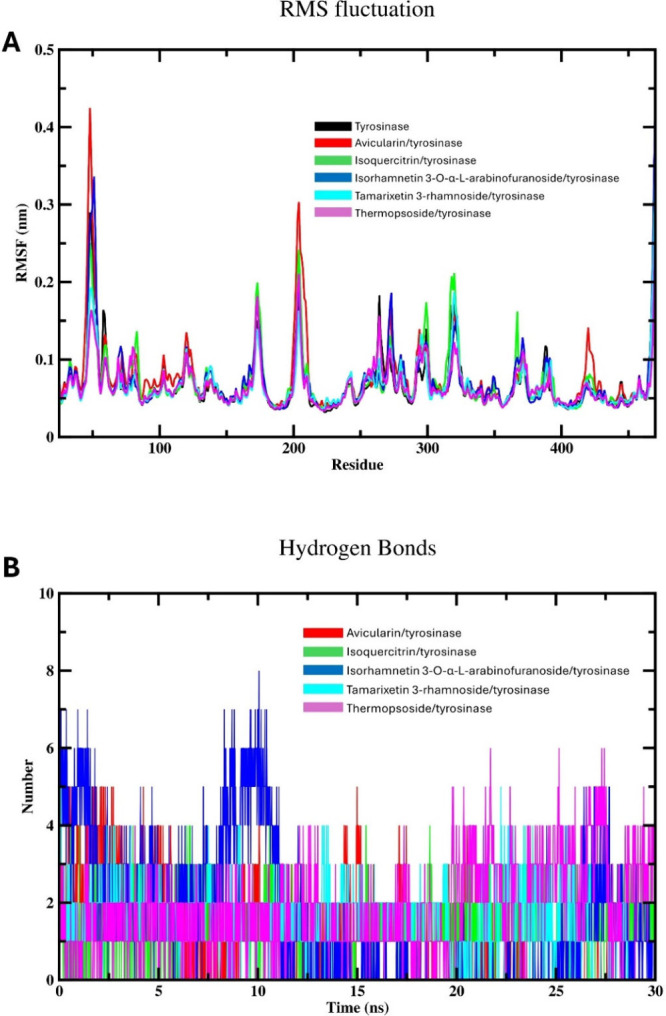
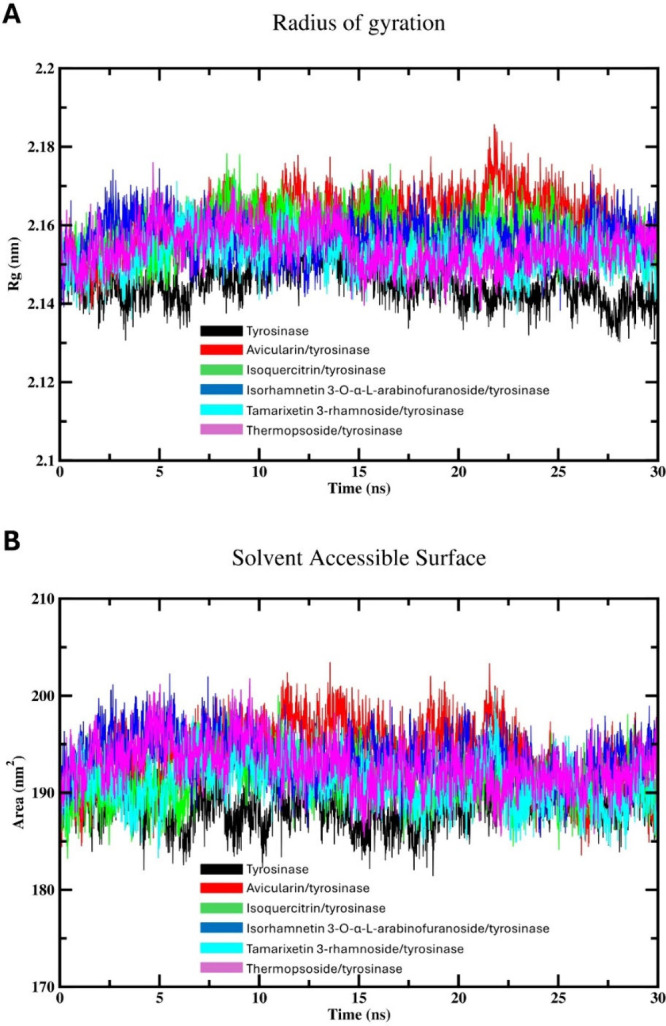
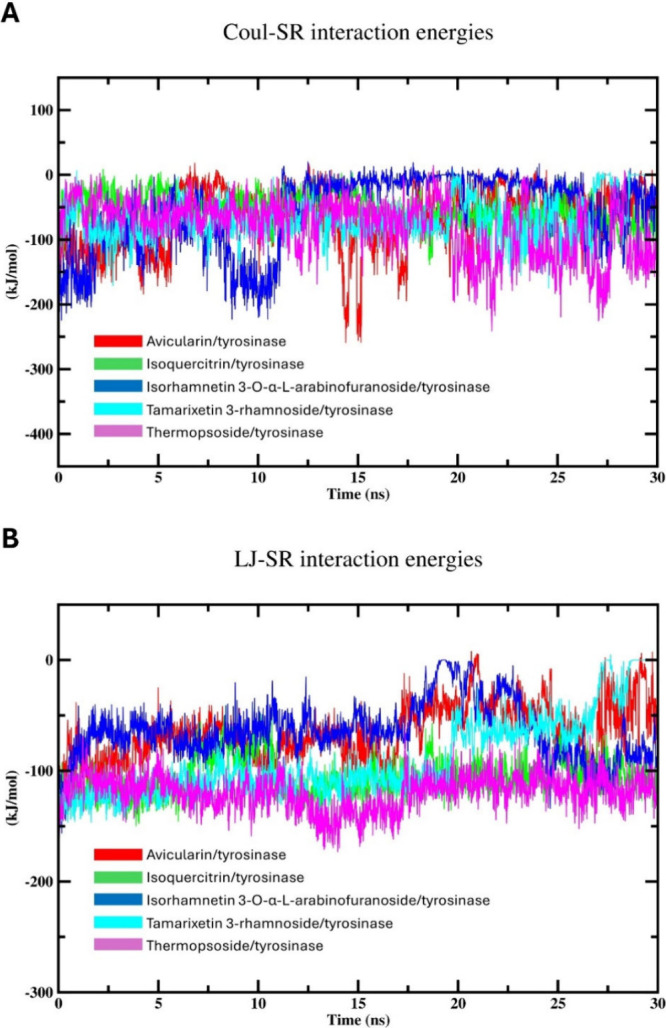
Flavonoids, natural compounds ubiquitous in the human diet, are esteemed for their multifaceted pharmacological properties. The tyrosinase inhibitory capacity of five flavonoids from was investigated through a comprehensive integration of and methodologies. Molecular docking simulations demonstrated the proficient binding of the isolated compounds to the principal binding site of tyrosinase, akin to the standard tyrosinase inhibitor kojic acid. These compounds exhibited analogous binding affinities, among which compound manifested notably heightened levels of polar bonds and hydrophobic interactions. Molecular dynamics (MD) simulations were utilized to explore the interaction dynamics between the isolated flavonoids and tyrosinase. The analysis of various MD parameters revealed consistent trajectories for the tested compounds, with compound demonstrating notable energy equilibration. Strong hydrogen bonding interactions were observed between the flavonoids and the tyrosinase active site. The results of interaction energy calculations showed a balanced interaction mediated by hydrophobic interactions, with compound exhibiting the lowest interaction energies. Additionally, the findings from MM/PBSA analysis demonstrated the lowest binding free energy for compound . Moreover, the tyrosinase inhibition assay revealed notable discrepancies among the studied flavonoids. Particularly, compound demonstrated the most pronounced anti-tyrosinase activity, as evidenced by its lowest IC value. This experimental outcome is consistent with the results of computational predictions. Therefore, flavonoids of might be valuable for the development of tyrosinase inhibitors.
黄酮类化合物是人类饮食中普遍存在的天然化合物,因其多方面的药理特性而受到重视。通过综合运用多种方法,对来自[具体来源未明确]的五种黄酮类化合物的酪氨酸酶抑制能力进行了研究。分子对接模拟表明,分离出的化合物能与酪氨酸酶的主要结合位点有效结合,类似于标准酪氨酸酶抑制剂曲酸。这些化合物表现出相似的结合亲和力,其中化合物[具体化合物未明确]表现出显著更高水平的极性键和疏水相互作用。利用分子动力学(MD)模拟来探索分离出的黄酮类化合物与酪氨酸酶之间的相互作用动力学。对各种MD参数的分析揭示了测试化合物的一致轨迹,化合物[具体化合物未明确]表现出显著的能量平衡。在黄酮类化合物与酪氨酸酶活性位点之间观察到了强烈的氢键相互作用。相互作用能计算结果表明,疏水相互作用介导了平衡的相互作用,化合物[具体化合物未明确]表现出最低的相互作用能。此外,MM/PBSA分析结果表明化合物[具体化合物未明确]的结合自由能最低。此外,酪氨酸酶抑制试验揭示了所研究的黄酮类化合物之间存在显著差异。特别是,化合物[具体化合物未明确]表现出最显著的抗酪氨酸酶活性,其最低IC值证明了这一点。这一实验结果与计算预测结果一致。因此,[具体来源未明确]的黄酮类化合物可能对酪氨酸酶抑制剂的开发具有重要价值。