Zhang Haihan, He Kevin, Li Zheng, Tsoi Lam C, Zhou Xiang
Department of Biostatistics, University of Michigan, Ann Arbor, Michigan, United States of America.
Department of Dermatology, University of Michigan Medical School, Ann Arbor, Michigan, United States of America.
PLoS Genet. 2024 Dec 2;20(12):e1011503. doi: 10.1371/journal.pgen.1011503. eCollection 2024 Dec.
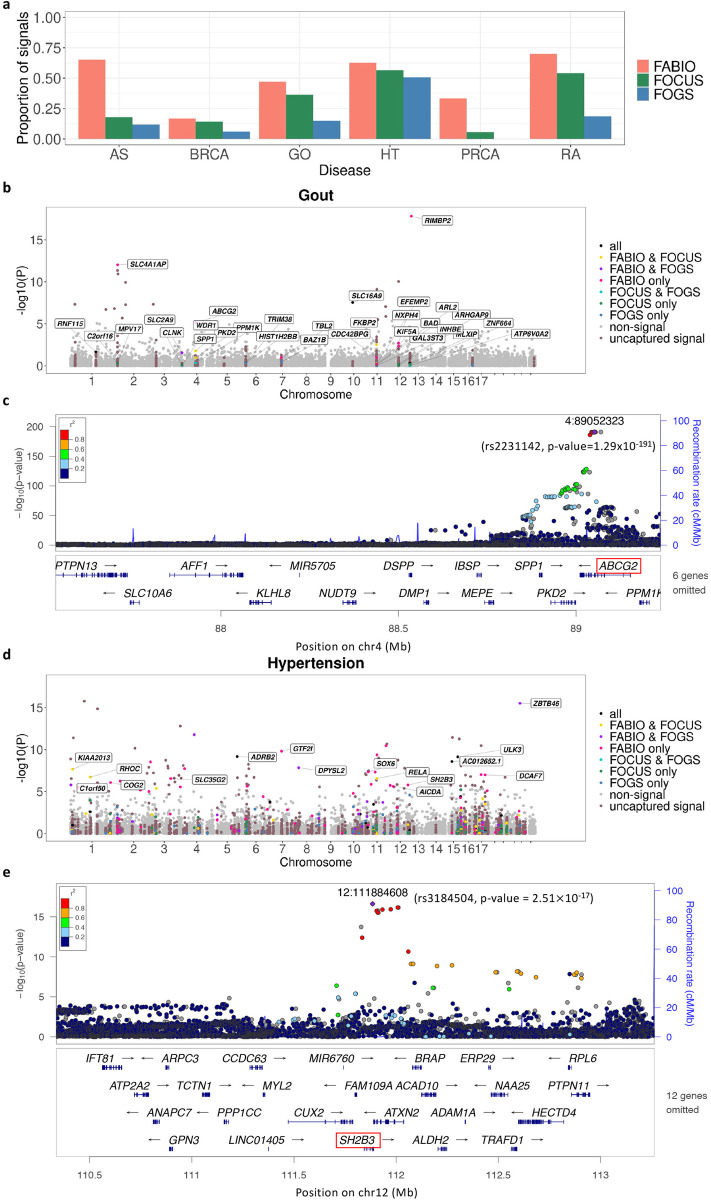
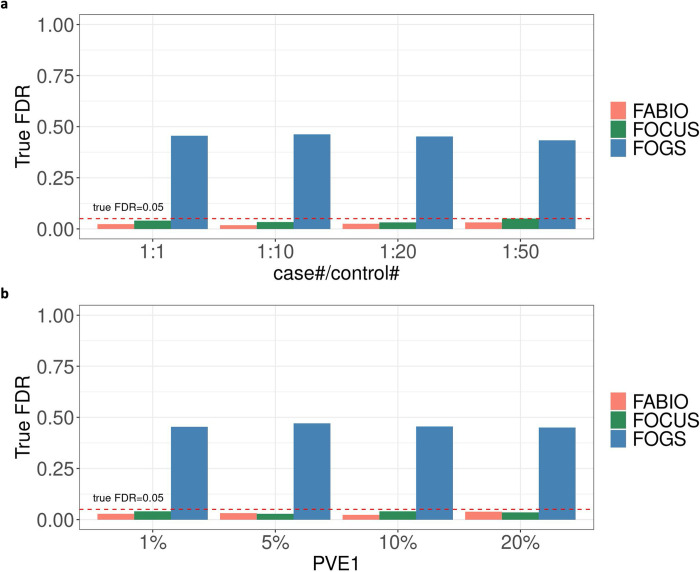
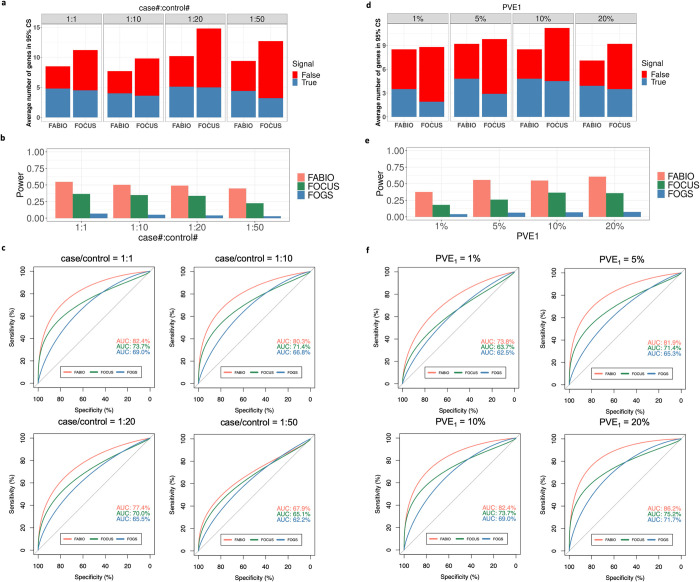
Transcriptome-wide association studies (TWAS) have emerged as a powerful tool for identifying gene-trait associations by integrating gene expression mapping studies with genome-wide association studies (GWAS). While most existing TWAS approaches focus on marginal analyses through examining one gene at a time, recent developments in TWAS fine-mapping methods enable the joint modeling of multiple genes to refine the identification of potentially causal ones. However, these fine-mapping methods have primarily focused on modeling quantitative traits and examining local genomic regions, leading to potentially suboptimal performance. Here, we present FABIO, a TWAS fine-mapping method specifically designed for binary traits that is capable of modeling all genes jointly on an entire chromosome. FABIO employs a probit model to directly link the genetically regulated expression (GReX) of genes to binary outcomes while taking into account the GReX correlation among all genes residing on a chromosome. As a result, FABIO effectively controls false discoveries while offering substantial power gains over existing TWAS fine-mapping approaches. We performed extensive simulations to evaluate the performance of FABIO and applied it for in-depth analyses of six binary disease traits in the UK Biobank. In the real datasets, FABIO significantly reduced the size of the causal gene sets by 27.9%-36.9% over existing approaches across traits. Leveraging its improved power, FABIO successfully prioritized multiple potentially causal genes associated with the diseases, including GATA3 for asthma, ABCG2 for gout, and SH2B3 for hypertension. Overall, FABIO represents an effective tool for TWAS fine-mapping of disease traits.
全转录组关联研究(TWAS)已成为一种强大的工具,通过整合基因表达图谱研究与全基因组关联研究(GWAS)来识别基因与性状之间的关联。虽然大多数现有的TWAS方法通过一次检查一个基因来专注于边际分析,但TWAS精细定位方法的最新进展使得能够对多个基因进行联合建模,以优化对潜在因果基因的识别。然而,这些精细定位方法主要集中在对数量性状进行建模和检查局部基因组区域,导致性能可能次优。在这里,我们提出了FABIO,这是一种专门为二元性状设计的TWAS精细定位方法,能够在整条染色体上对所有基因进行联合建模。FABIO采用概率单位模型,在考虑位于一条染色体上的所有基因的基因调控表达(GReX)相关性的同时,直接将基因的GReX与二元结果联系起来。因此,FABIO有效地控制了错误发现,同时比现有的TWAS精细定位方法具有显著的功效提升。我们进行了广泛的模拟来评估FABIO的性能,并将其应用于英国生物银行中六种二元疾病性状的深入分析。在真实数据集中,与现有方法相比,FABIO在各性状上显著减少了因果基因集的大小,降幅为27.9%-36.9%。利用其提高的功效,FABIO成功地对与疾病相关的多个潜在因果基因进行了优先排序,包括哮喘相关的GATA3、痛风相关的ABCG2和高血压相关的SH2B3。总体而言,FABIO是疾病性状TWAS精细定位的有效工具。