De Debajyoti, Thapliyal Nemika, Prakash Tiwari Ved, Toyama Yuki, Flemming Hansen D, Kay Lewis E, Vallurupalli Pramodh
Tata Institute of Fundamental Research Hyderabad, Ranga Reddy District, Hyderabad 500046, India.
Department of Molecular Genetics, University of Toronto, Toronto M5S 1A8, Canada.
Proc Natl Acad Sci U S A. 2024 Dec 10;121(50):e2416682121. doi: 10.1073/pnas.2416682121. Epub 2024 Dec 4.
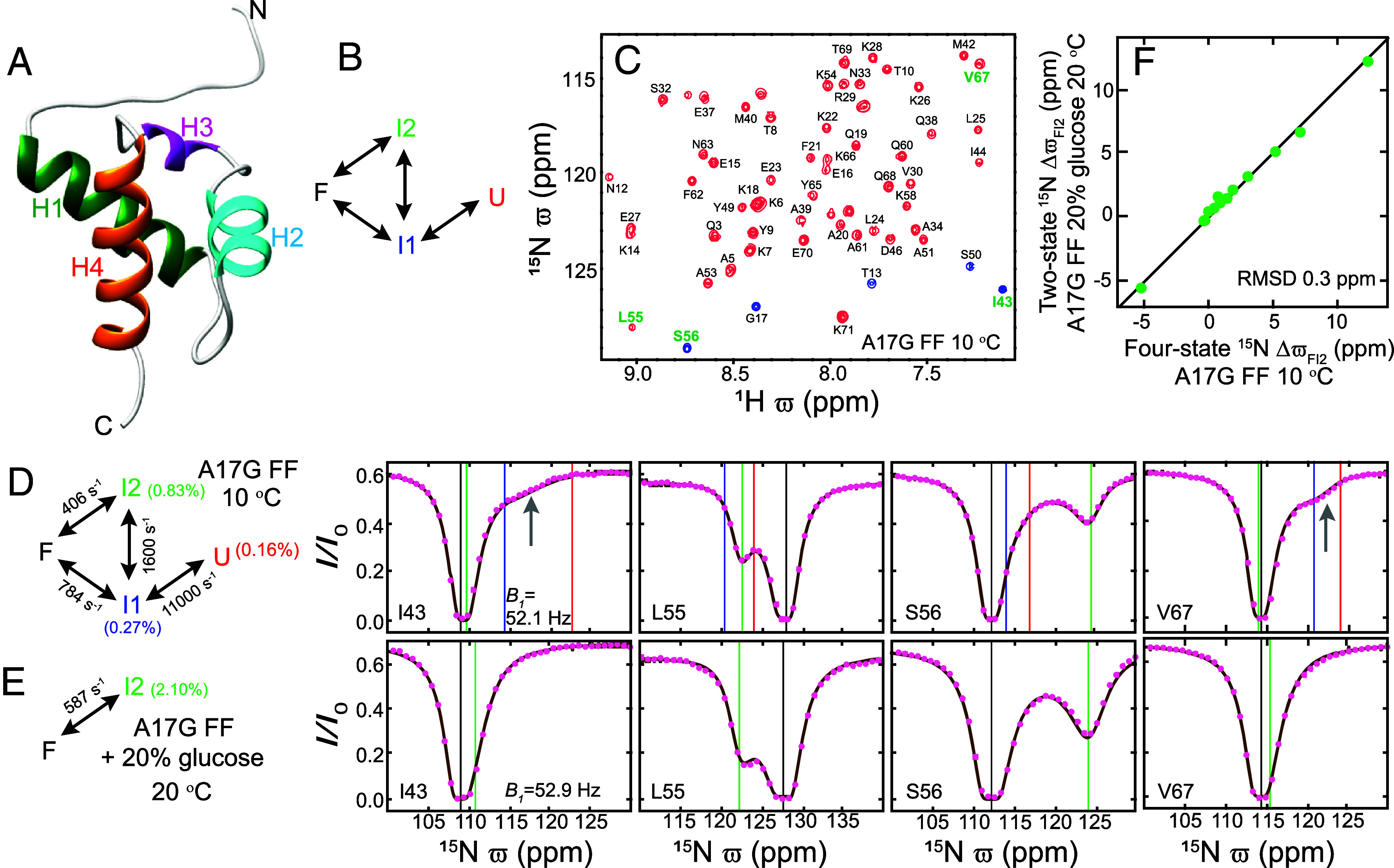
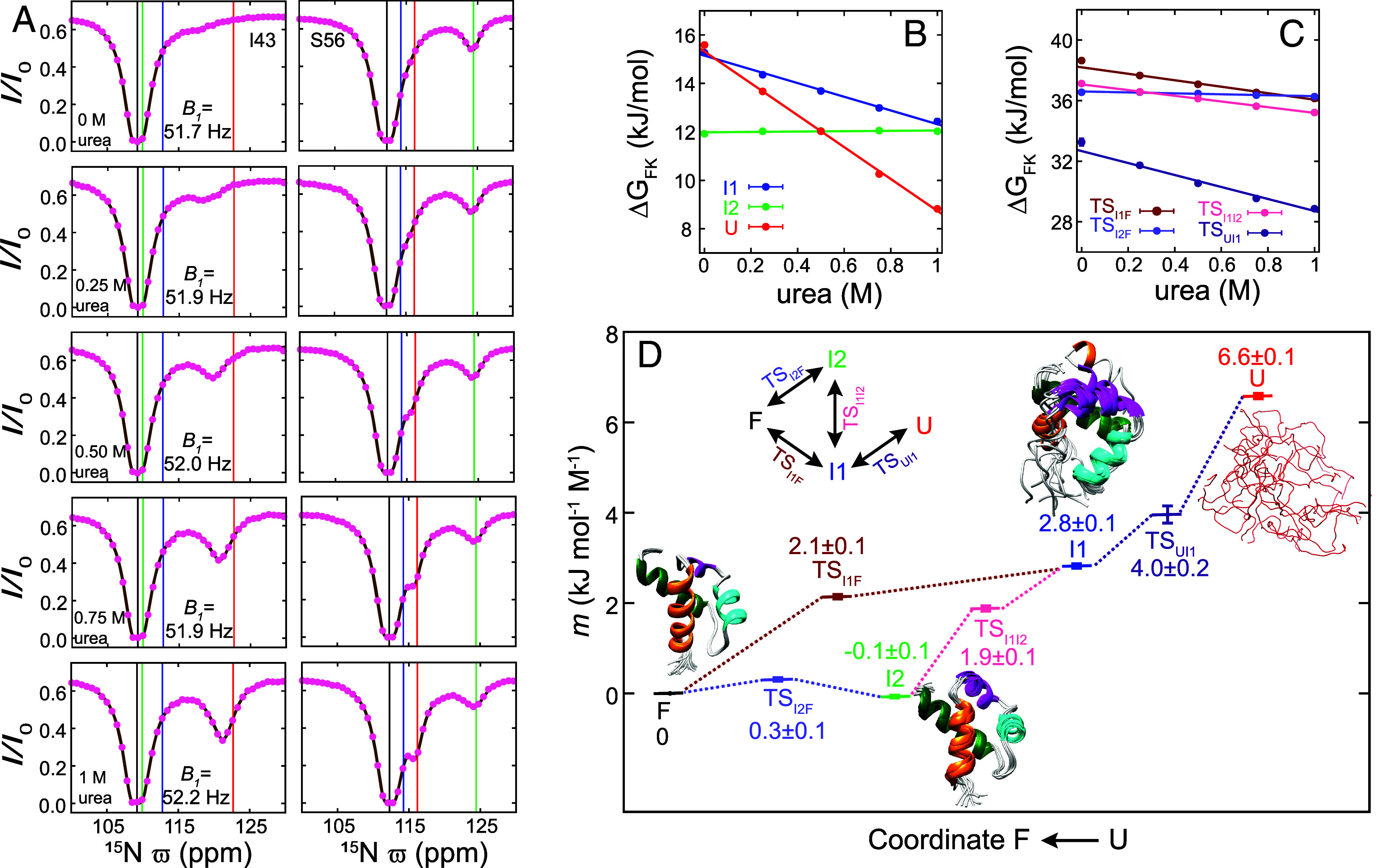
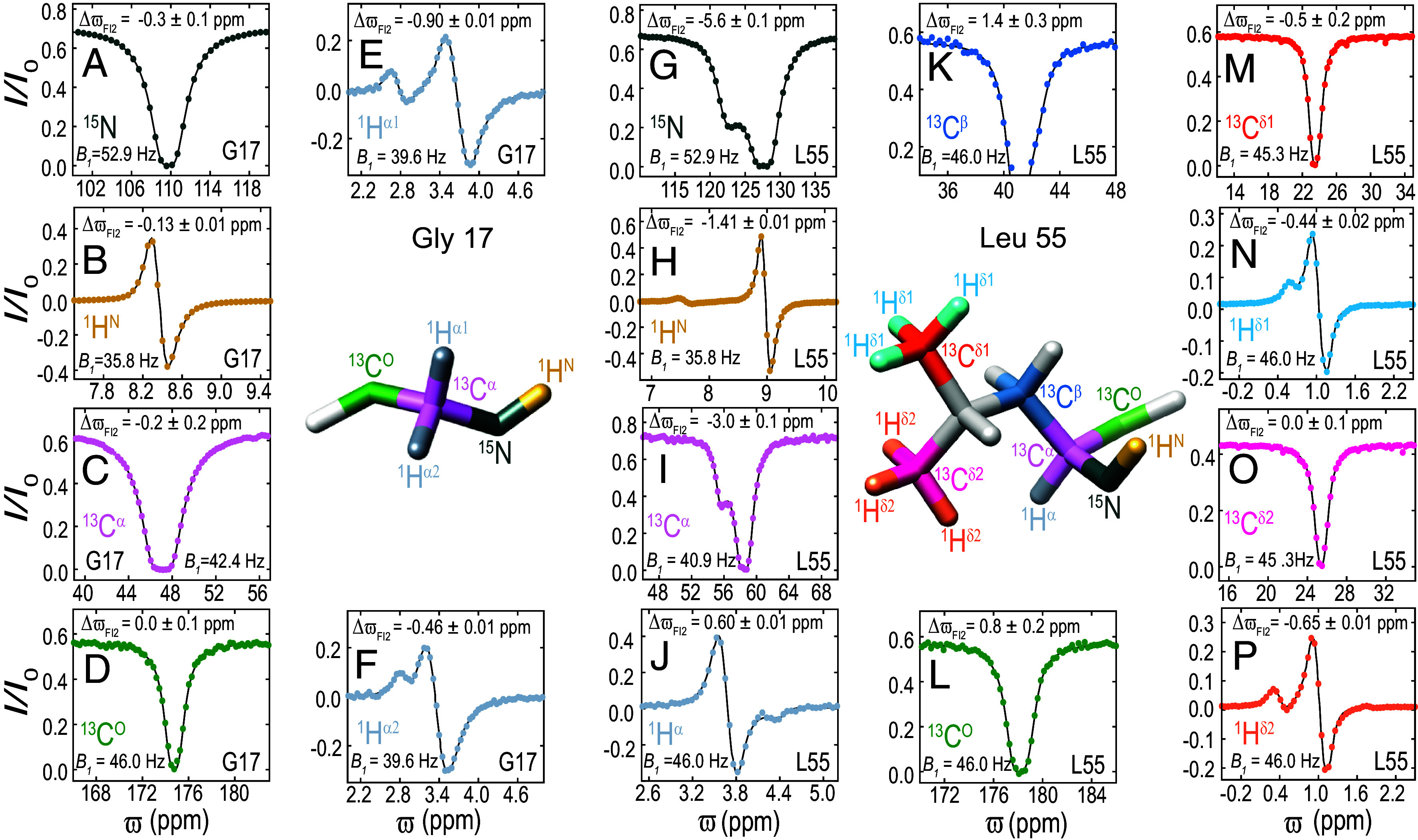
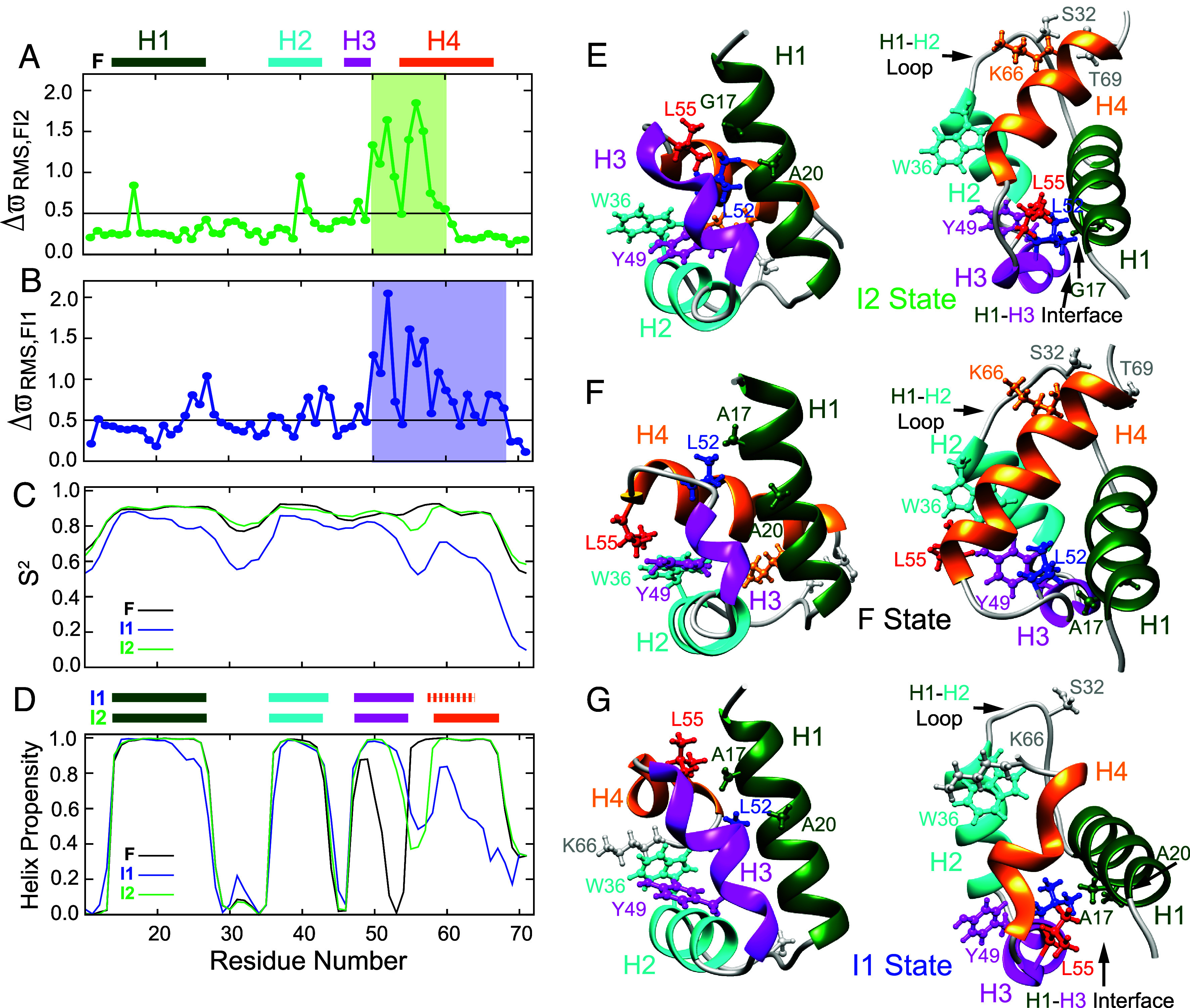
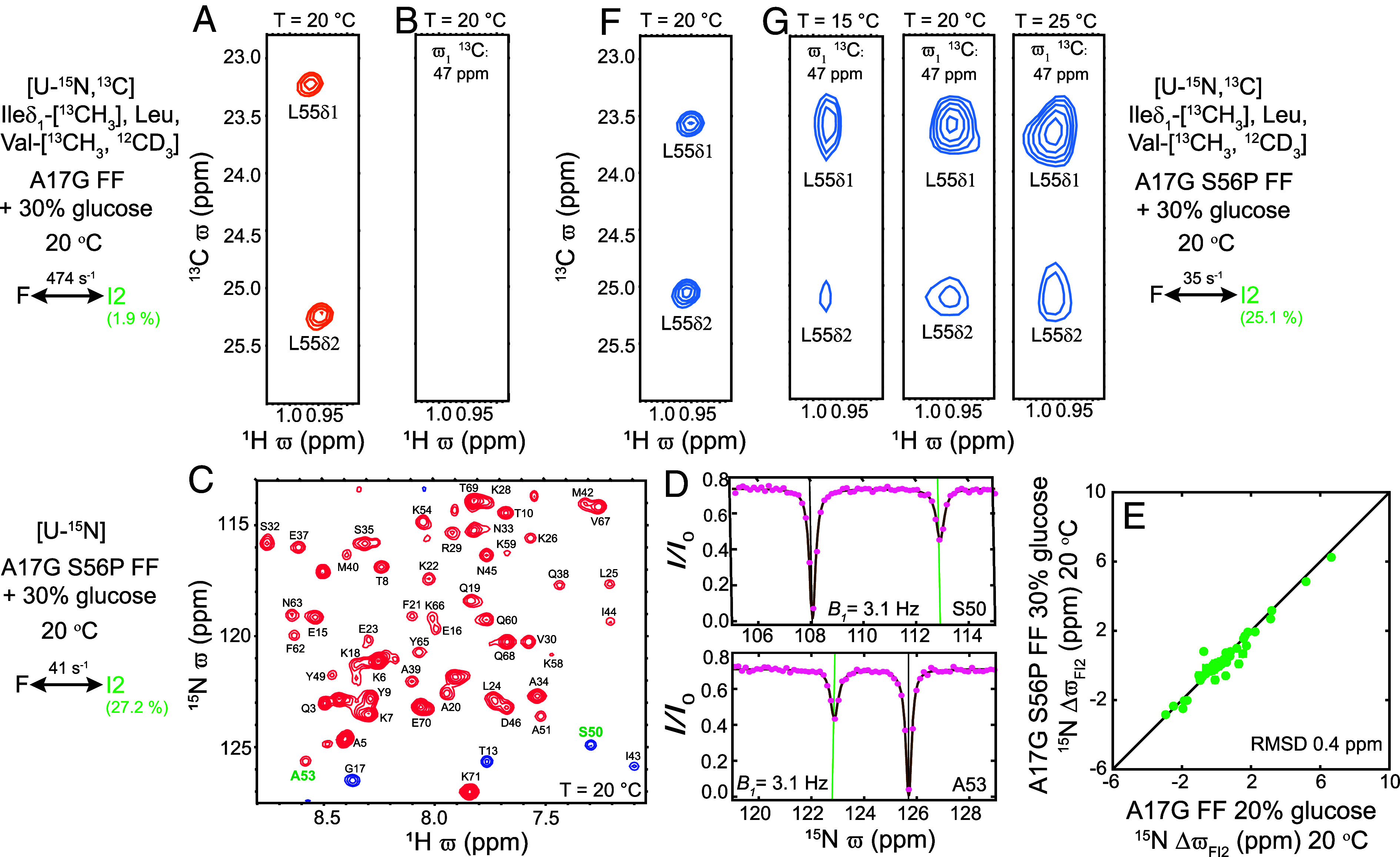
Despite the tremendous accomplishments of AlpaFold2/3 in predicting biomolecular structure, the protein folding problem remains unsolved in the sense that accurate atomistic models of how protein molecules fold into their native conformations from an unfolded ensemble are still elusive. Here, using chemical exchange saturation transfer (CEST) NMR experiments and a comprehensive four-state kinetic model of the folding trajectory of a 71 residue four-helix bundle FF domain from human HYPA/FBP11 we present an atomic resolution structure of a transiently formed intermediate, I2, that along with the structure of a second intermediate, I1, provides a description of the FF domain folding trajectory. By recording CEST profiles as a function of urea concentration the extent of compaction along the folding pathway is evaluated. Our data establish that unlike the partially disordered I1 state, the I2 intermediate that is also formed before the rate-limiting folding barrier is well ordered and compact like the native conformer, while retaining nonnative interactions similar to those found in I1. The slow-interconversion from I2 to F, involving changes in secondary structure and the breaking of nonnative interactions, proceeds via a compact transition-state. Interestingly, the native state of the FF1 domain from human p190-A Rho GAP resembles the I2 conformation, suggesting that well-ordered folding intermediates can be repurposed by nature in structurally related proteins to assume functional roles. It is anticipated that the strategy for elucidation of sparsely populated and transiently formed structures of intermediates along kinetic pathways described here will be of use in other studies of protein dynamics.
尽管AlpaFold2/3在预测生物分子结构方面取得了巨大成就,但蛋白质折叠问题在某种意义上仍然没有得到解决,即关于蛋白质分子如何从未折叠的集合体折叠成其天然构象的精确原子模型仍然难以捉摸。在这里,我们使用化学交换饱和转移(CEST)核磁共振实验以及来自人类HYPA/FBP11的71个残基的四螺旋束FF结构域折叠轨迹的综合四态动力学模型,展示了一个瞬时形成的中间体I2的原子分辨率结构,该中间体与第二个中间体I1的结构一起,描述了FF结构域的折叠轨迹。通过记录作为尿素浓度函数的CEST图谱,评估了沿着折叠途径的压缩程度。我们的数据表明,与部分无序的I1状态不同,在限速折叠屏障之前也形成的I2中间体像天然构象一样有序且紧凑,同时保留了与I1中发现的类似的非天然相互作用。从I2到F的缓慢相互转化,涉及二级结构的变化和非天然相互作用的断裂,通过一个紧凑的过渡态进行。有趣的是,来自人类p190 - A Rho GAP的FF1结构域的天然状态类似于I2构象,这表明自然界可以在结构相关的蛋白质中重新利用有序的折叠中间体来承担功能角色。预计这里描述的用于阐明沿着动力学途径的中间体的稀疏分布和瞬时形成结构的策略将在蛋白质动力学的其他研究中有用。