Hu Frank, Chen Michael S, Rotskoff Grant M, Kanan Matthew W, Markland Thomas E
Department of Chemistry, Stanford University, Stanford, California 94305, United States.
Simons Center for Computational Physical Chemistry, Department of Chemistry, New York University, New York, New York 10003, United States.
ACS Cent Sci. 2024 Nov 13;10(11):2162-2170. doi: 10.1021/acscentsci.4c01132. eCollection 2024 Nov 27.
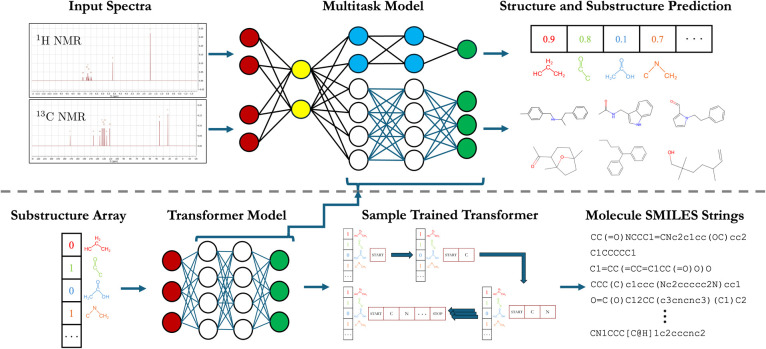
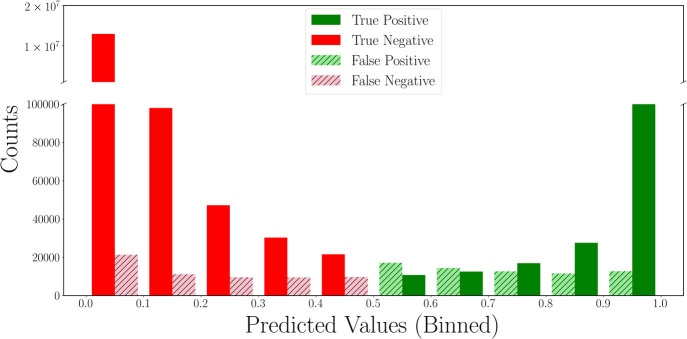
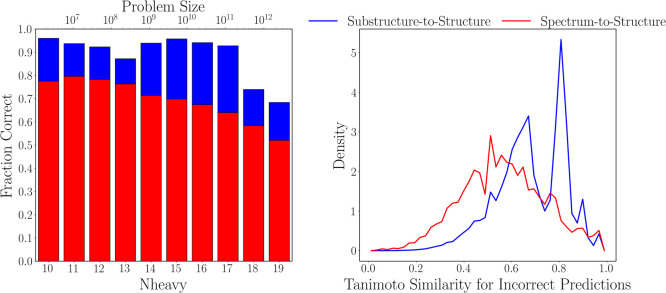
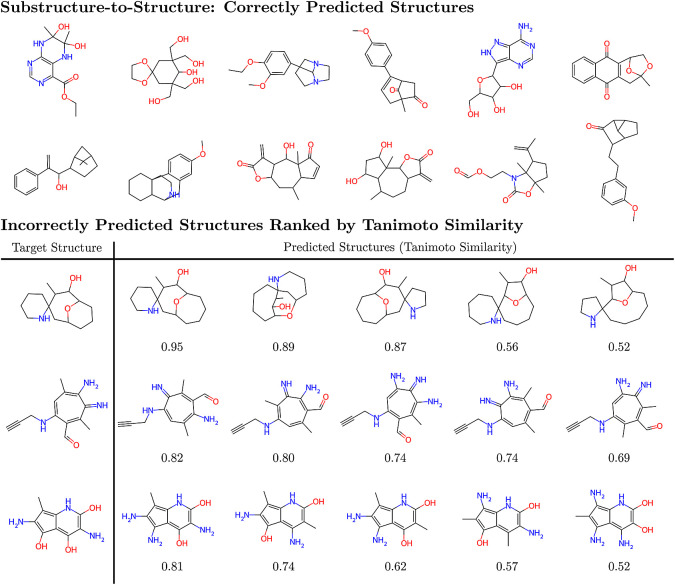
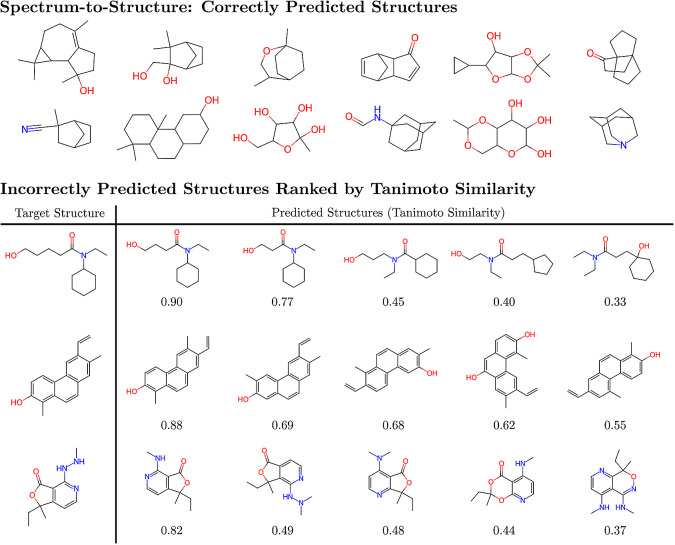
Rapid determination of molecular structures can greatly accelerate workflows across many chemical disciplines. However, elucidating structure using only one-dimensional (1D) NMR spectra, the most readily accessible data, remains an extremely challenging problem because of the combinatorial explosion of the number of possible molecules as the number of constituent atoms is increased. Here, we introduce a multitask machine learning framework that predicts the molecular structure (formula and connectivity) of an unknown compound solely based on its 1D H and/or C NMR spectra. First, we show how a transformer architecture can be constructed to efficiently solve the task, traditionally performed by chemists, of assembling large numbers of molecular fragments into molecular structures. Integrating this capability with a convolutional neural network, we build an end-to-end model for predicting structure from spectra that is fast and accurate. We demonstrate the effectiveness of this framework on molecules with up to 19 heavy (non-hydrogen) atoms, a size for which there are trillions of possible structures. Without relying on any prior chemical knowledge such as the molecular formula, we show that our approach predicts the exact molecule 69.6% of the time within the first 15 predictions, reducing the search space by up to 11 orders of magnitude.
快速确定分子结构能够极大地加速许多化学学科的工作流程。然而,仅使用一维(1D)核磁共振谱(最容易获取的数据)来阐明结构,仍然是一个极具挑战性的问题,因为随着组成原子数量的增加,可能分子的数量会呈组合式爆炸增长。在此,我们引入了一个多任务机器学习框架,该框架仅基于未知化合物的一维氢谱和/或碳谱来预测其分子结构(分子式和连接性)。首先,我们展示了如何构建一种变压器架构,以有效地解决传统上由化学家执行的将大量分子片段组装成分子结构的任务。将此能力与卷积神经网络相结合,我们构建了一个从光谱预测结构的端到端模型,该模型快速且准确。我们在含有多达19个重(非氢)原子的分子上证明了该框架的有效性,对于这种规模的分子,可能的结构有万亿种。在不依赖任何先验化学知识(如分子式)的情况下,我们表明我们的方法在前15次预测中,有69.6%的时间能预测出确切的分子,将搜索空间减少了多达11个数量级。