Ekholm Andreas, Wang Yinxi, Vallon-Christersson Johan, Boissin Constance, Rantalainen Mattias
Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Box 281, Stockholm, 171 77, Sweden.
Division of Oncology, Department of Clinical Sciences Lund, Lund University, Lund, Sweden.
BMC Cancer. 2024 Dec 11;24(1):1510. doi: 10.1186/s12885-024-13248-9.
In breast cancer, several gene expression assays have been developed to provide a more personalised treatment. This study focuses on the prediction of two molecular proliferation signatures: an 11-gene proliferation score and the MKI67 proliferation marker gene. The aim was to assess whether these could be predicted from digital whole slide images (WSIs) using deep learning models.
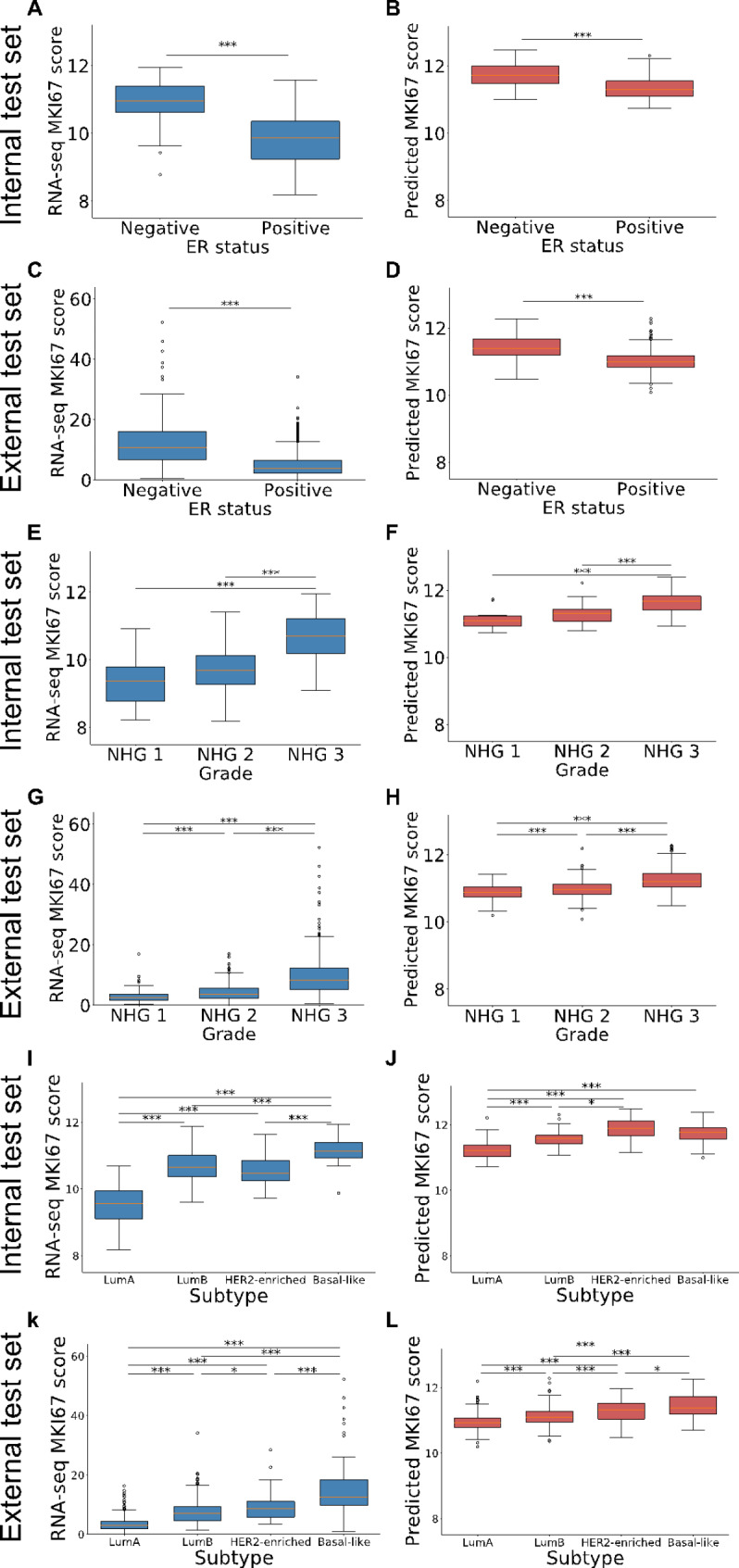
WSIs and RNA-sequencing data from 819 invasive breast cancer patients were included for training, and models were evaluated on an internal test set of 172 cases as well as on 997 cases from a fully independent external test set. Two deep Convolutional Neural Network (CNN) models were optimised using WSIs and gene expression readouts from RNA-sequencing data of either the proliferation signature or the proliferation marker, and assessed using Spearman correlation (r). Prognostic performance was assessed through Cox proportional hazard modelling, estimating hazard ratios (HR).
Optimised CNNs successfully predicted the proliferation score and proliferation marker on the unseen internal test set (ρ = 0.691(p < 0.001) with R = 0.438, and ρ = 0.564 (p < 0.001) with R = 0.251 respectively) and on the external test set (ρ = 0.502 (p < 0.001) with R = 0.319, and ρ = 0.403 (p < 0.001) with R = 0.222 respectively). Patients with a high proliferation score or marker were significantly associated with a higher risk of recurrence or death in the external test set (HR = 1.65 (95% CI: 1.05-2.61) and HR = 1.84 (95% CI: 1.17-2.89), respectively).
The results from this study suggest that gene expression levels of proliferation scores can be predicted directly from breast cancer morphology in WSIs using CNNs and that the predictions provide prognostic information that could be used in research as well as in the clinical setting.
在乳腺癌中,已开发出多种基因表达检测方法以提供更个性化的治疗。本研究聚焦于两种分子增殖特征的预测:一种11基因增殖评分和MKI67增殖标记基因。目的是评估能否使用深度学习模型从数字全切片图像(WSIs)中预测这些特征。
纳入819例浸润性乳腺癌患者的WSIs和RNA测序数据用于训练,并在172例的内部测试集以及来自完全独立外部测试集的997例病例上对模型进行评估。使用来自增殖特征或增殖标记的RNA测序数据的WSIs和基因表达读数优化了两个深度卷积神经网络(CNN)模型,并使用斯皮尔曼相关性(r)进行评估。通过Cox比例风险模型评估预后性能,估计风险比(HR)。
优化后的CNN在未见的内部测试集(分别为ρ = 0.691(p < 0.001),R = 0.438,以及ρ = 0.564(p < 0.001),R = 0.251)和外部测试集(分别为ρ = 0.502(p < 0.001),R = 0.319,以及ρ = 0.403(p < 0.001),R = 0.222)上成功预测了增殖评分和增殖标记。在外部测试集中,增殖评分或标记高的患者与复发或死亡风险较高显著相关(分别为HR = 1.65(95% CI:1.05 - 2.61)和HR = 1.84(95% CI:1.17 - 2.89))。
本研究结果表明,使用CNN可直接从WSIs中的乳腺癌形态预测增殖评分的基因表达水平,且这些预测提供的预后信息可用于研究及临床环境。