Shen Wan Xiang, Cui Chao, Su Xiaorui, Zhang Zaixi, Velez-Arce Alejandro, Wang Jianming, Shi Xiangcheng, Zhang Yanbing, Wu Jie, Chen Yu Zong, Zitnik Marinka
Department of Biomedical Informatics, Harvard Medical School, Boston, MA, USA.
Department of Chemistry, National University of Singapore, 117543, Singapore.
Res Sq. 2024 Dec 4:rs.3.rs-2988283. doi: 10.21203/rs.3.rs-2988283/v2.
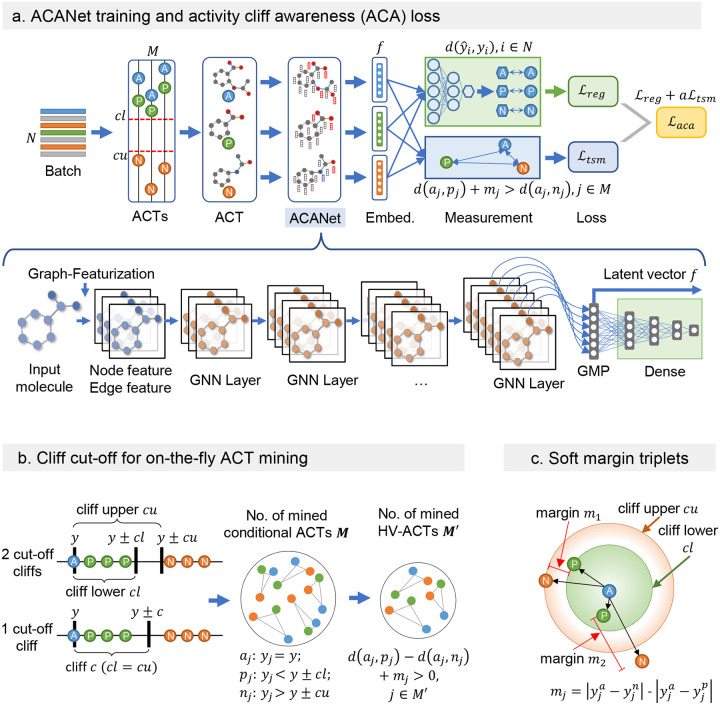
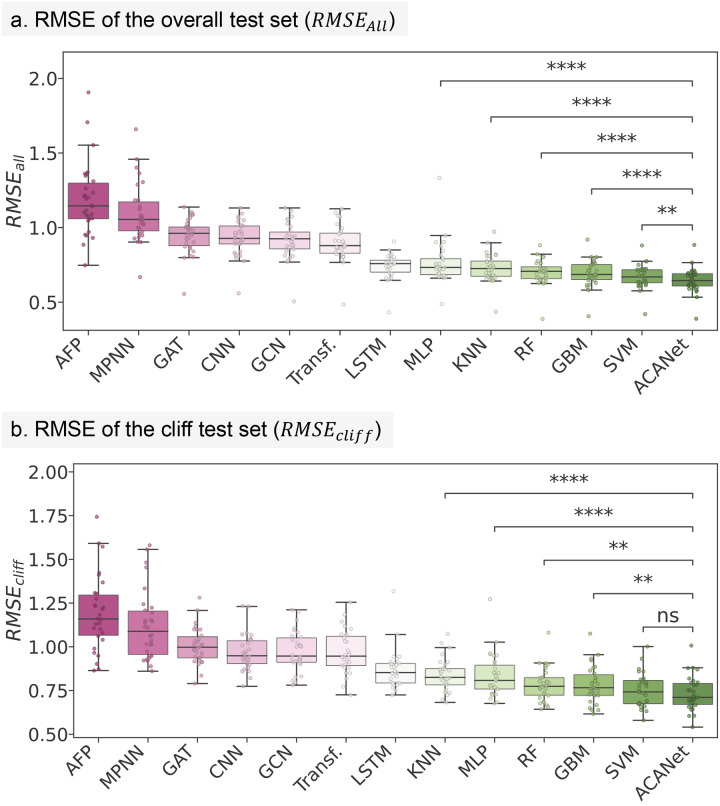
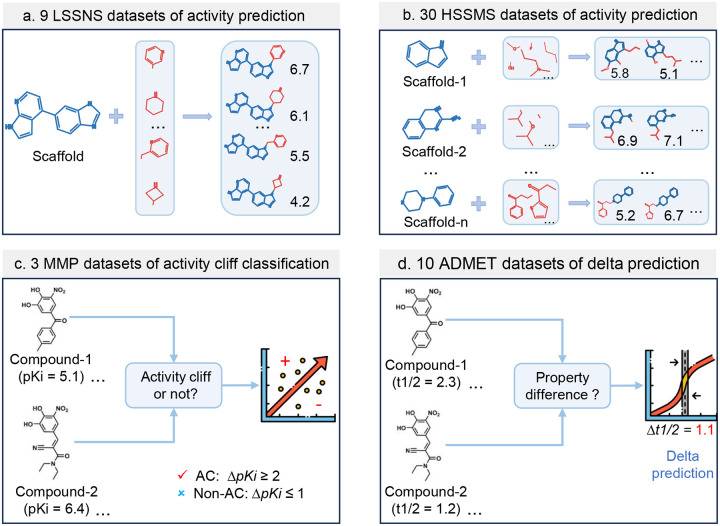
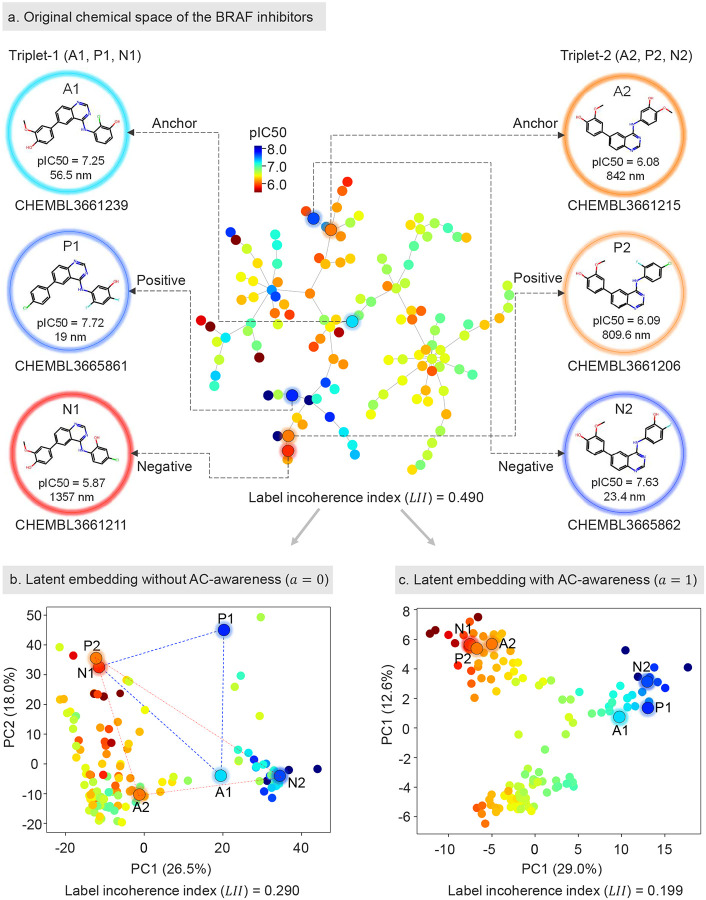
Modeling molecular activity and quantitative structure-activity relationships of chemical compounds is critical in drug design. Graph neural networks, which utilize molecular structures as frames, have shown success in assessing the biological activity of chemical compounds, guiding the selection and optimization of candidates for further development. However, current models often overlook activity cliffs (ACs)-cases where structurally similar molecules exhibit different bioactivities-due to latent spaces primarily optimized for structural features. Here, we introduce AC-awareness (ACA), an inductive bias designed to enhance molecular representation learning for activity modeling. The ACA jointly optimizes metric learning in the latent space and task performance in the target space, making models more sensitive to ACs. We develop ACANet, an AC-informed contrastive learning approach that can be integrated with any graph neural network. Experiments on 39 benchmark datasets demonstrate that AC-informed representations of chemical compounds consistently outperform standard models in bioactivity prediction across both regression and classification tasks. AC-informed models show strong performance in predicting pharmacokinetic and safety-relevant molecular properties. ACA paves the way toward activity-informed molecular representations, providing a valuable tool for the early stages of lead compound identification, refinement, and virtual screening.
对化合物的分子活性和定量构效关系进行建模在药物设计中至关重要。以分子结构为框架的图神经网络在评估化合物的生物活性、指导进一步开发的候选物的选择和优化方面已取得成功。然而,由于潜在空间主要针对结构特征进行优化,当前模型常常忽略活性悬崖(ACs),即结构相似的分子表现出不同生物活性的情况。在此,我们引入了AC感知(ACA),这是一种归纳偏差,旨在增强用于活性建模的分子表示学习。ACA在潜在空间中联合优化度量学习和目标空间中的任务性能,使模型对ACs更加敏感。我们开发了ACANet,一种可与任何图神经网络集成的基于AC的对比学习方法。在39个基准数据集上进行的实验表明,在回归和分类任务的生物活性预测中,化合物的基于AC的表示始终优于标准模型。基于AC的模型在预测药代动力学和安全相关分子性质方面表现出强大性能。ACA为基于活性的分子表示铺平了道路,为先导化合物识别、优化和虚拟筛选的早期阶段提供了有价值的工具。