Chang Wei-Shan, Harvey Erin, Mahar Jackie E, Firth Cadhla, Shi Mang, Simon-Loriere Etienne, Geoghegan Jemma L, Wille Michelle
School of Medical Sciences, The University of Sydney, Sydney, NSW, Australia.
Health and Biosecurity, Commonwealth Scientific and Industrial Research Organisation, Canberra, ACT, Australia.
Commun Biol. 2024 Dec 20;7(1):1687. doi: 10.1038/s42003-024-07212-3.
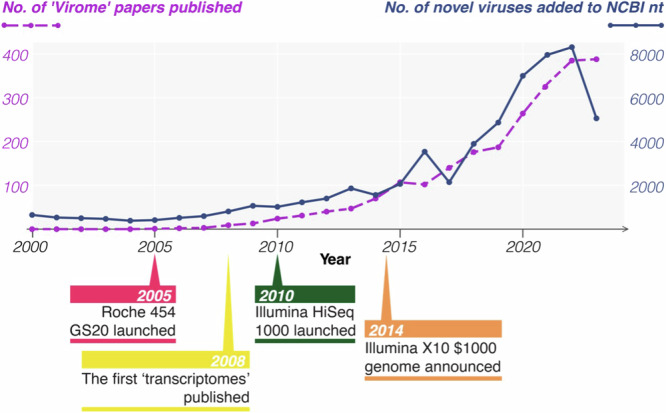
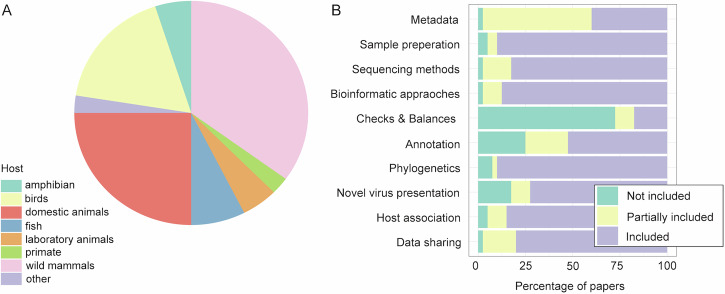
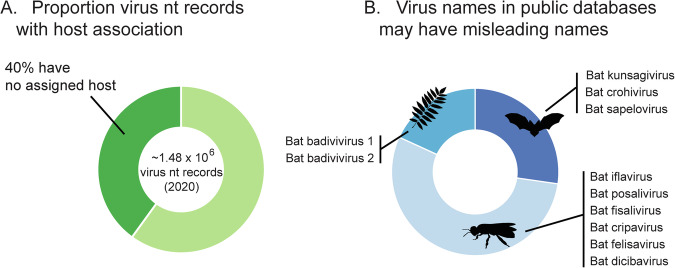
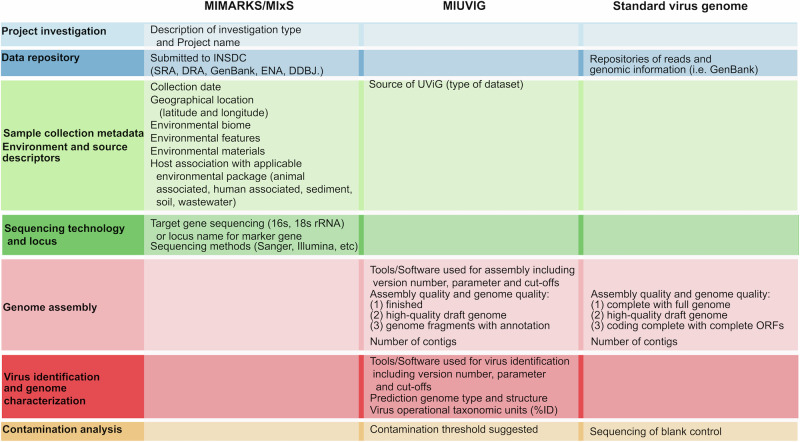
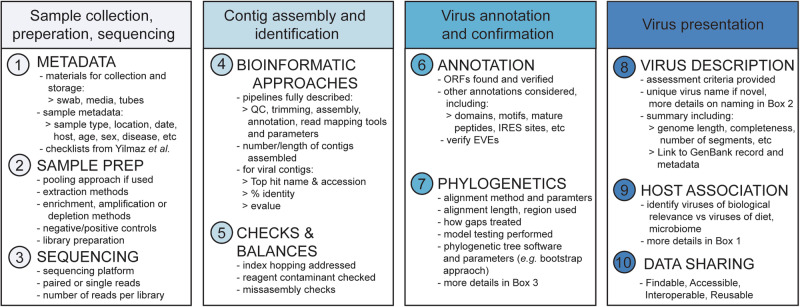
Over the last decade metagenomic sequencing has facilitated an increasing number of virome-scale studies, leading to an exponential expansion in understanding of virus diversity. This is partially driven by the decreasing costs of metagenomic sequencing, improvements in computational tools for revealing novel viruses, and an increased understanding of the key role that viruses play in human and animal health. A central concern associated with this remarkable increase in the number of virome-scale studies is the lack of broadly accepted "gold standards" for reporting the data and results generated. This is of particular importance for animal virome studies as there are a multitude of nuanced approaches for both data presentation and analysis, all of which impact the resulting outcomes. As such, the results of published studies can be difficult to contextualise and may be of reduced utility due to reporting deficiencies. Herein, we aim to address these reporting issues by outlining recommendations for the presentation of virome data, encouraging a transparent communication of findings that can be interpreted in evolutionary and ecological contexts.
在过去十年中,宏基因组测序推动了越来越多病毒组规模的研究,从而在对病毒多样性的理解上呈指数级扩展。这部分得益于宏基因组测序成本的降低、用于发现新型病毒的计算工具的改进,以及对病毒在人类和动物健康中所起关键作用的进一步认识。与病毒组规模研究数量的显著增加相关的一个核心问题是,缺乏广泛认可的用于报告所产生数据和结果的“金标准”。这对于动物病毒组研究尤为重要,因为在数据呈现和分析方面有多种细微差别不同的方法,所有这些都会影响最终结果。因此,已发表研究的结果可能难以进行背景化解读,并且由于报告缺陷其效用可能会降低。在此,我们旨在通过概述病毒组数据呈现的建议来解决这些报告问题,鼓励以透明的方式传达研究结果,以便在进化和生态背景下进行解读。