The Globe Institute, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark.
Max Planck Tandem Group in Computational Biology, Department of Biological Sciences, Universidad de los Andes, Bogota, Colombia.
PLoS Comput Biol. 2023 Aug 28;19(8):e1011422. doi: 10.1371/journal.pcbi.1011422. eCollection 2023 Aug.
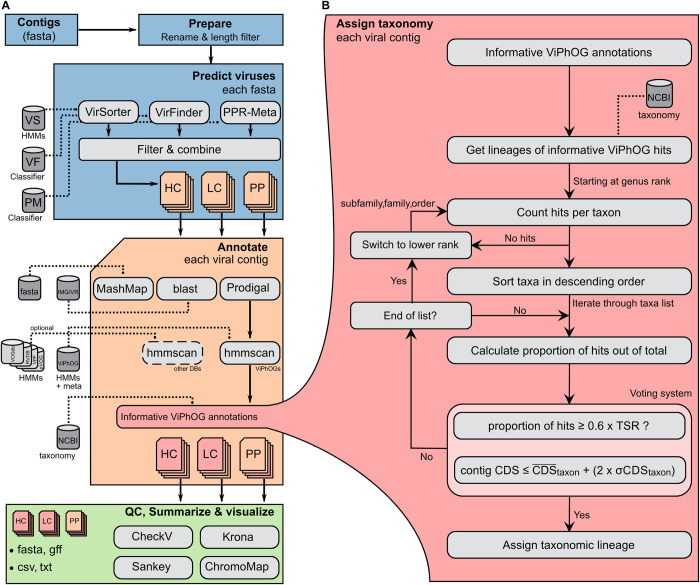
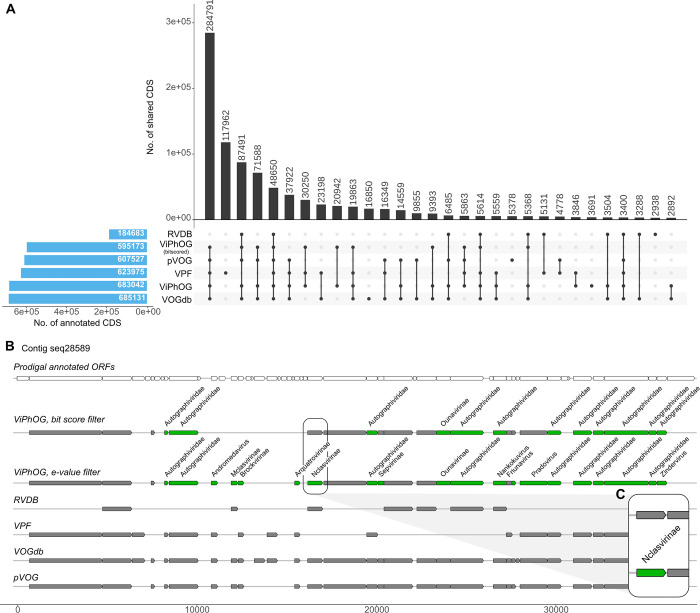
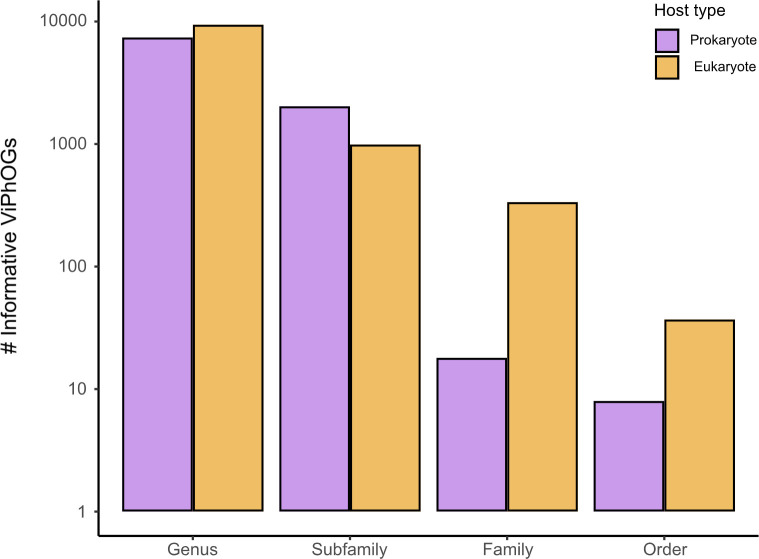
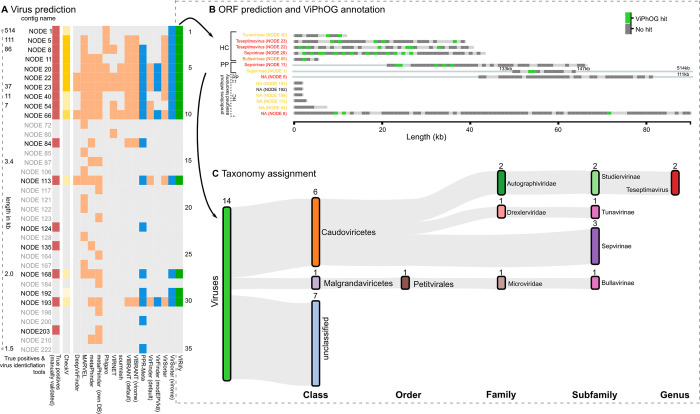
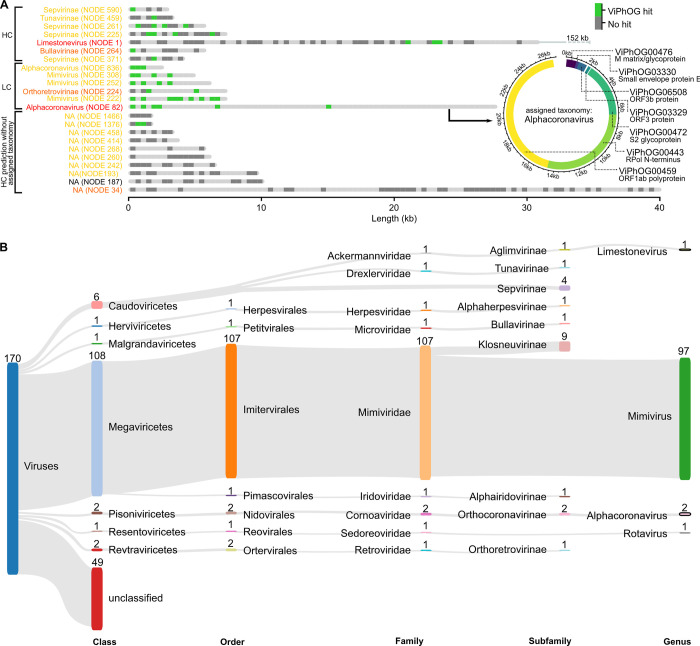
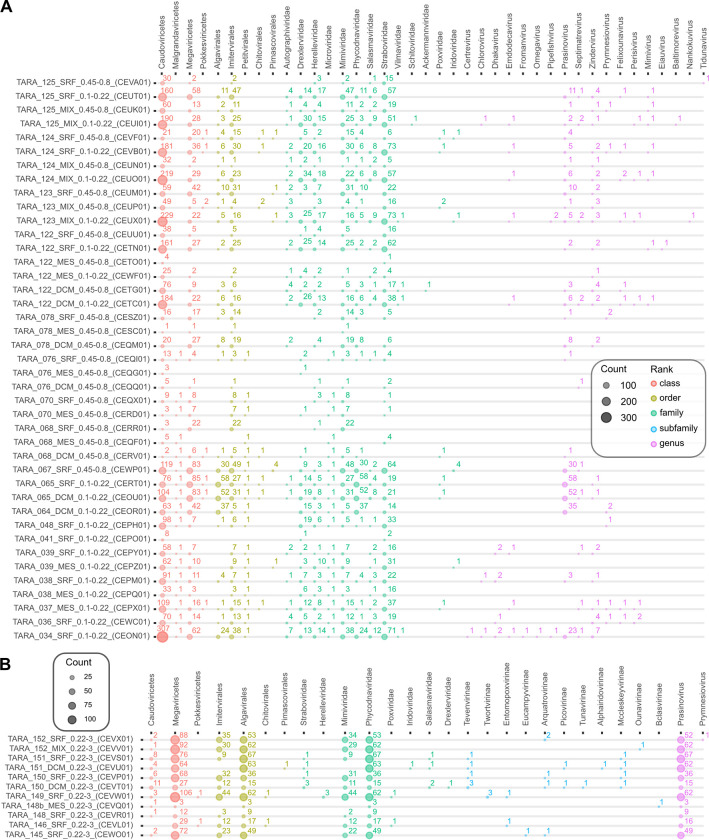
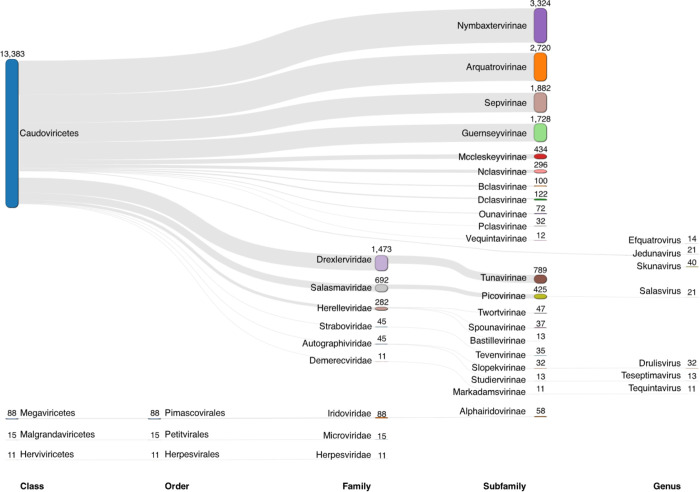
The study of viral communities has revealed the enormous diversity and impact these biological entities have on various ecosystems. These observations have sparked widespread interest in developing computational strategies that support the comprehensive characterisation of viral communities based on sequencing data. Here we introduce VIRify, a new computational pipeline designed to provide a user-friendly and accurate functional and taxonomic characterisation of viral communities. VIRify identifies viral contigs and prophages from metagenomic assemblies and annotates them using a collection of viral profile hidden Markov models (HMMs). These include our manually-curated profile HMMs, which serve as specific taxonomic markers for a wide range of prokaryotic and eukaryotic viral taxa and are thus used to reliably classify viral contigs. We tested VIRify on assemblies from two microbial mock communities, a large metagenomics study, and a collection of publicly available viral genomic sequences from the human gut. The results showed that VIRify could identify sequences from both prokaryotic and eukaryotic viruses, and provided taxonomic classifications from the genus to the family rank with an average accuracy of 86.6%. In addition, VIRify allowed the detection and taxonomic classification of a range of prokaryotic and eukaryotic viruses present in 243 marine metagenomic assemblies. Finally, the use of VIRify led to a large expansion in the number of taxonomically classified human gut viral sequences and the improvement of outdated and shallow taxonomic classifications. Overall, we demonstrate that VIRify is a novel and powerful resource that offers an enhanced capability to detect a broad range of viral contigs and taxonomically classify them.
病毒群落的研究揭示了这些生物实体在各种生态系统中所具有的巨大多样性和影响力。这些观察结果引发了人们广泛的兴趣,促使开发出基于测序数据支持全面描述病毒群落的计算策略。在这里,我们介绍了 VIRify,这是一种新的计算管道,旨在为病毒群落提供用户友好且准确的功能和分类特征描述。VIRify 从宏基因组组装中识别病毒 contigs 和原噬菌体,并使用一系列病毒特征隐马尔可夫模型 (HMM) 对其进行注释。这些包括我们手动 curated 的特征 HMM,它们是广泛的原核和真核病毒分类群的特定分类标记,因此可用于可靠地对病毒 contigs 进行分类。我们在两个微生物模拟群落的组装体、一个大型宏基因组研究以及人类肠道中公开可用的病毒基因组序列集合上测试了 VIRify。结果表明,VIRify 可以识别出原核和真核病毒的序列,并提供从属到科的分类学分类,平均准确率为 86.6%。此外,VIRify 还可以检测和分类 243 个海洋宏基因组组装体中存在的一系列原核和真核病毒。最后,使用 VIRify 导致分类学上分类的人类肠道病毒序列数量大幅增加,并改进了过时和浅层的分类学分类。总体而言,我们证明 VIRify 是一种新颖而强大的资源,提供了增强的检测广泛的病毒 contigs 并对其进行分类学分类的能力。