Cancino-Muñoz Irving, Mulet-Bayona Juan Vicente, Salvador-García Carme, Tormo-Palop Nuria, Guna Remedios, Gimeno-Cardona Concepción, González-Candelas Fernando
Unidad Mixta Infección y Salud Pública FISABIO-Universidad de Valencia, Valencia, Spain.
Instituto de Biología Integrativa de Sistemas, I2SysBio (CSIC-UV), Valencia, Spain.
mBio. 2025 Feb 5;16(2):e0316424. doi: 10.1128/mbio.03164-24. Epub 2024 Dec 27.
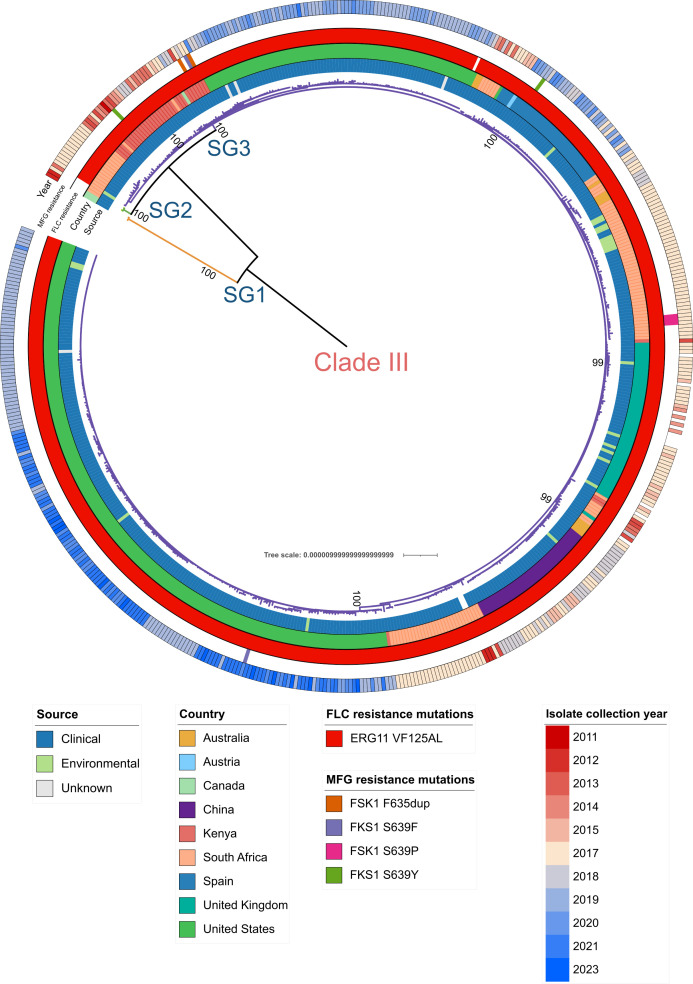
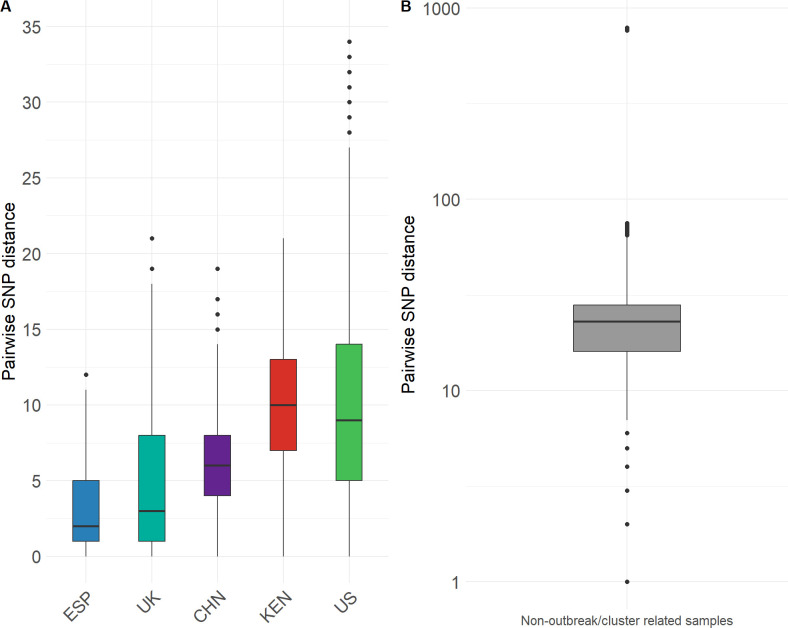
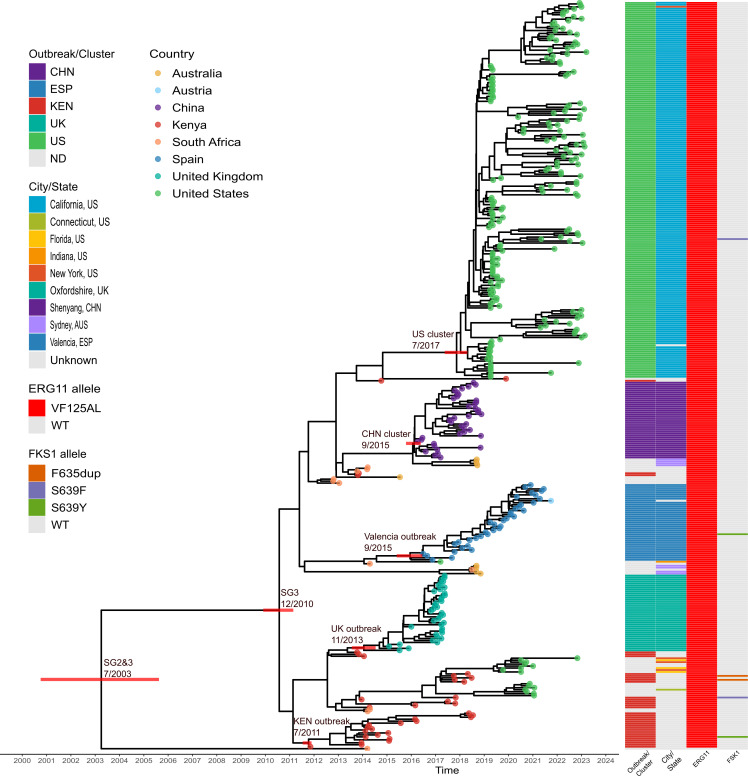
The rapid increase in infections caused by the emerging fungal pathogen is of global concern, and understanding its expansion is a priority. The phylogenetic diversity of the yeast is clustered in five major clades, among which clade III is particularly relevant, as most of its strains exhibit resistance to fluconazole, reducing the therapeutic alternatives and provoking outbreaks that are difficult to control. In this study, we have investigated the phylogenetic structure of clade III by analyzing a global collection of 566 genomes. We have identified three subgroups within clade III, among which two are genetically most closely related. Moreover, we have estimated the evolutionary rate of clade III to be 2.25e-7 s/s/y (2.87 changes per year). We found that one of these subgroups shows intrinsic resistance to fluconazole and is responsible for the majority of cases within this clade globally. We inferred that this subgroup may have originated around December 2010 (95% High Probability Density (HPD): April 2010-June 2011), and since then it has spread across continents, generating multiple large outbreaks, each with a unique pattern of transmission and dissemination. These results highlight the remarkable ability of the pathogen to adapt to its environment and its rapid global spread, underscoring the urgent need to address this epidemiological challenge effectively.IMPORTANCEThe number of cases affected by has increased worryingly worldwide. Among the currently recognized clades, clade III has the highest proportion of fluconazole-resistant cases and is spreading very rapidly, causing large nosocomial outbreaks across the globe. By analyzing complete fungal genomes from around the world, we have confirmed the origin of this clade and unraveled its dispersal patterns in the early 2010s. This finding provides knowledge that may be helpful to the public health authorities for the control of the disease.
这种新出现的真菌病原体导致的感染迅速增加,引起了全球关注,了解其传播情况是当务之急。该酵母的系统发育多样性聚集在五个主要分支中,其中分支III尤为重要,因为其大多数菌株对氟康唑耐药,减少了治疗选择,并引发了难以控制的疫情。在本研究中,我们通过分析566个全球基因组样本,研究了分支III的系统发育结构。我们在分支III中鉴定出三个亚组,其中两个在遗传上关系最为密切。此外,我们估计分支III的进化速率为2.25e-7 s/s/y(每年2.87个变化)。我们发现其中一个亚组对氟康唑具有固有抗性,并且在全球该分支内的大多数病例中占主导地位。我们推断这个亚组可能起源于2010年12月左右(95%高概率密度(HPD):2010年4月 - 2011年6月),从那时起它已蔓延至各大洲,引发了多次大规模疫情,每次疫情都有独特的传播和扩散模式。这些结果凸显了该病原体适应环境的显著能力及其在全球的快速传播,强调了有效应对这一流行病学挑战的迫切需求。重要性在全球范围内,受[病原体名称]影响的病例数量增长令人担忧。在目前公认的分支中,分支III的氟康唑耐药病例比例最高,并且传播非常迅速,在全球范围内导致了大量医院感染爆发。通过分析来自世界各地的完整真菌基因组,我们确定了这个分支的起源,并揭示了其在21世纪10年代初的传播模式。这一发现为公共卫生当局控制该疾病提供了可能有用的信息。