Heidari Razieh, Assadollahi Vahideh, Marashi Seyedeh Negar, Elahian Fatemeh, Mirzaei Seyed Abbas
Cancer Research Center, Basic Health Sciences Institute, Shahrekord University of Medical Sciences, Shahrekord, Iran.
Department of Medical Biotechnology, School of Advanced Technologies, Shahrekord University of Medical Sciences, Shahrekord, Iran.
Cancer Rep (Hoboken). 2025 Jan;8(1):e70098. doi: 10.1002/cnr2.70098.
Bioinformatics analysis of hepatocellular carcinoma (HCC) expression profiles can aid in understanding its molecular mechanisms and identifying new targets for diagnosis and treatment.
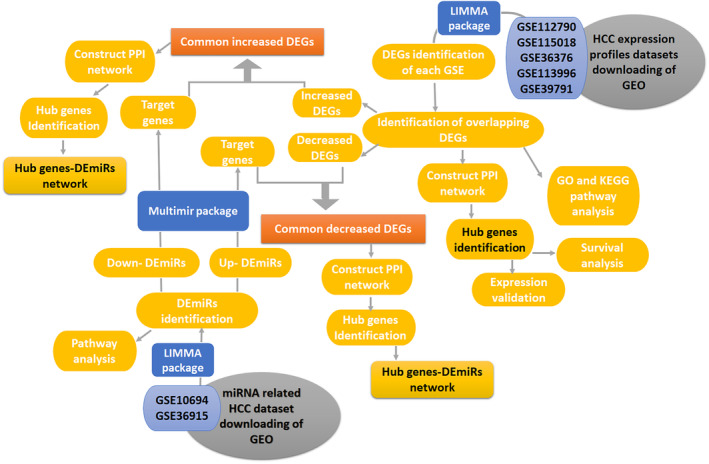
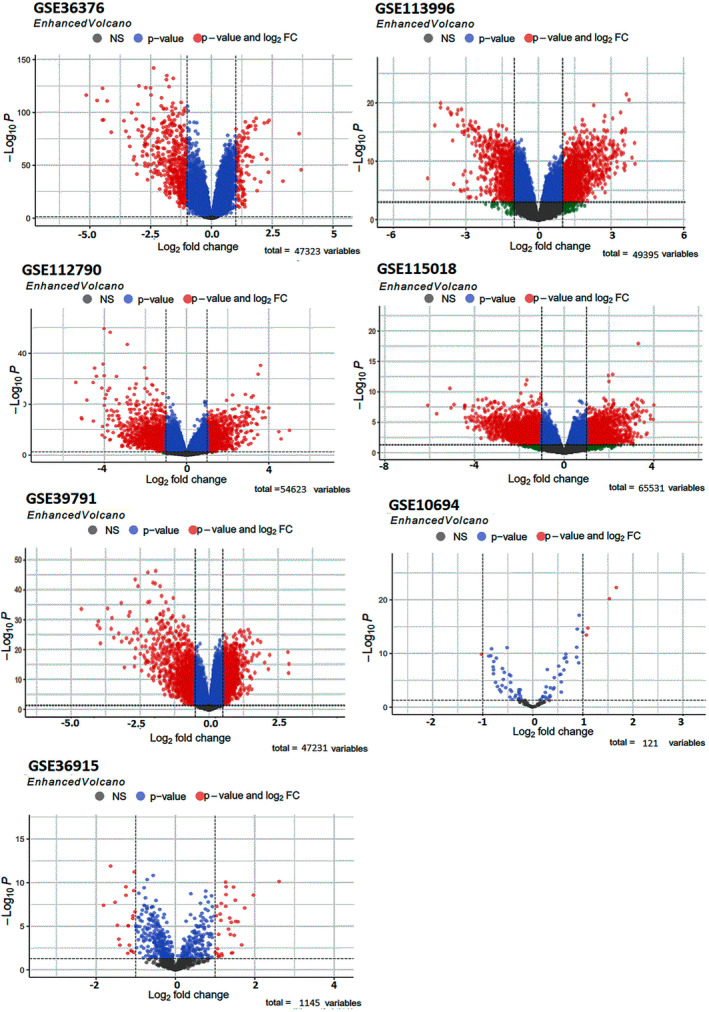
In this study, we analyzed expression profile datasets and miRNA expression profiles related to HCC from the GEO using R software to detect differentially expressed genes (DEGs) and differentially expressed miRNAs (DEmiRs).
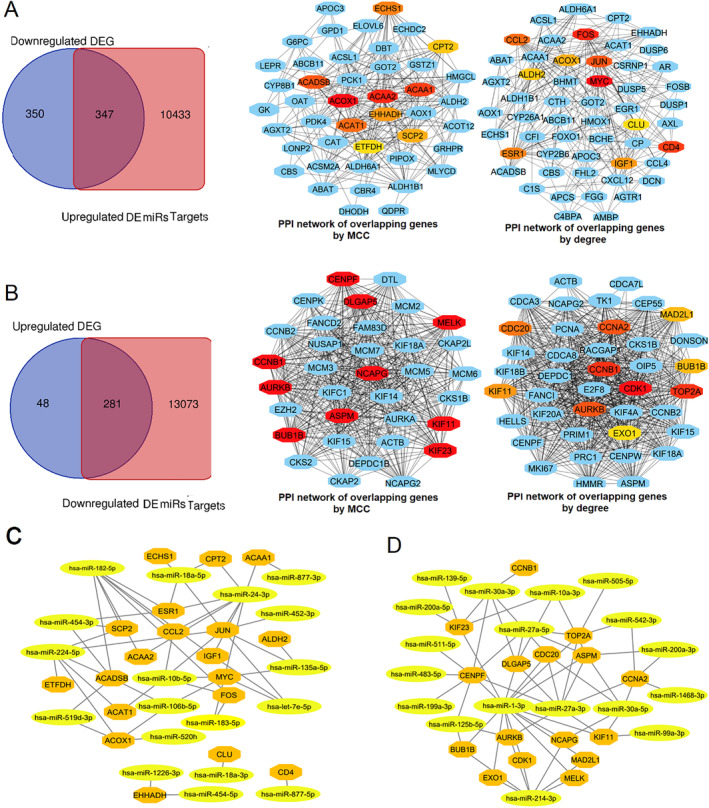
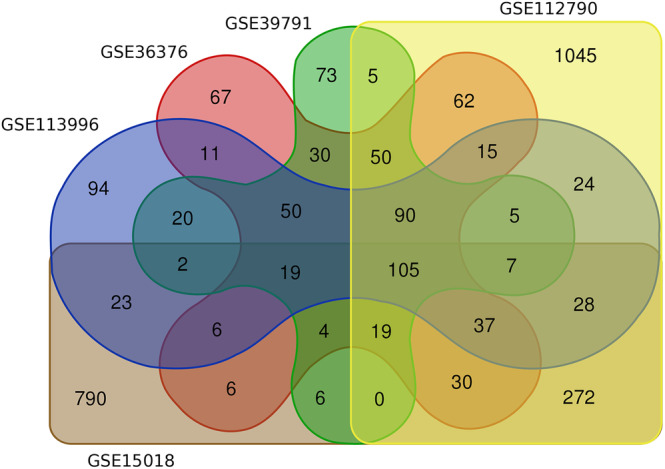
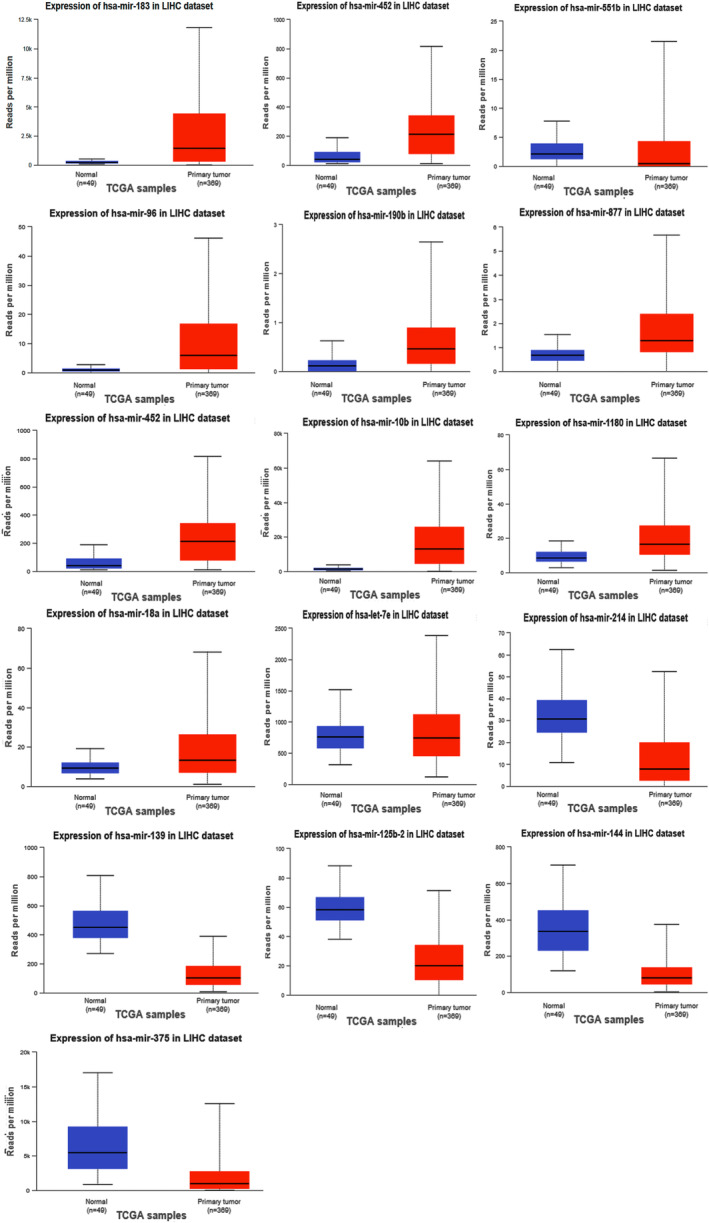
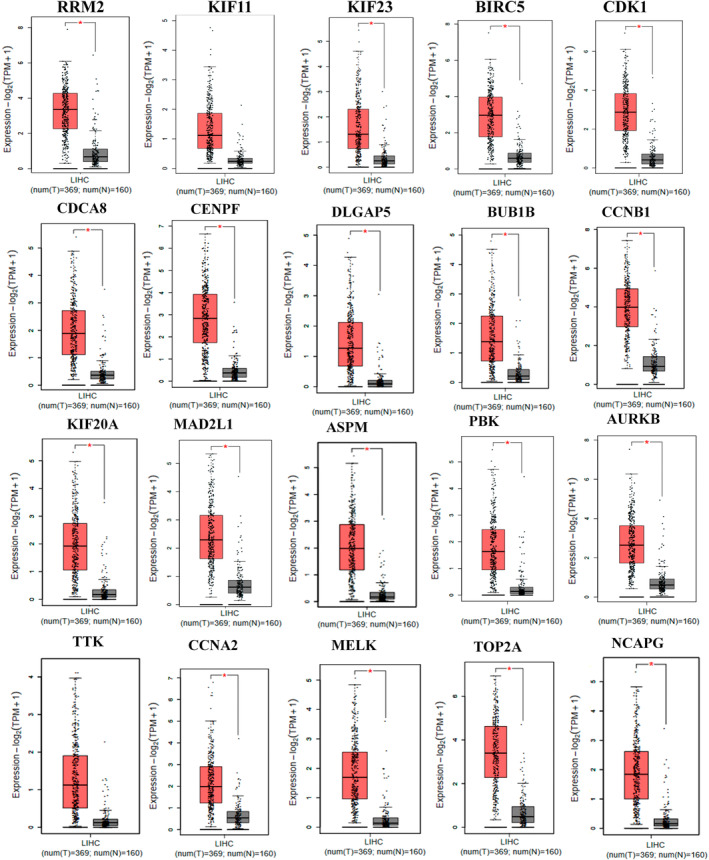
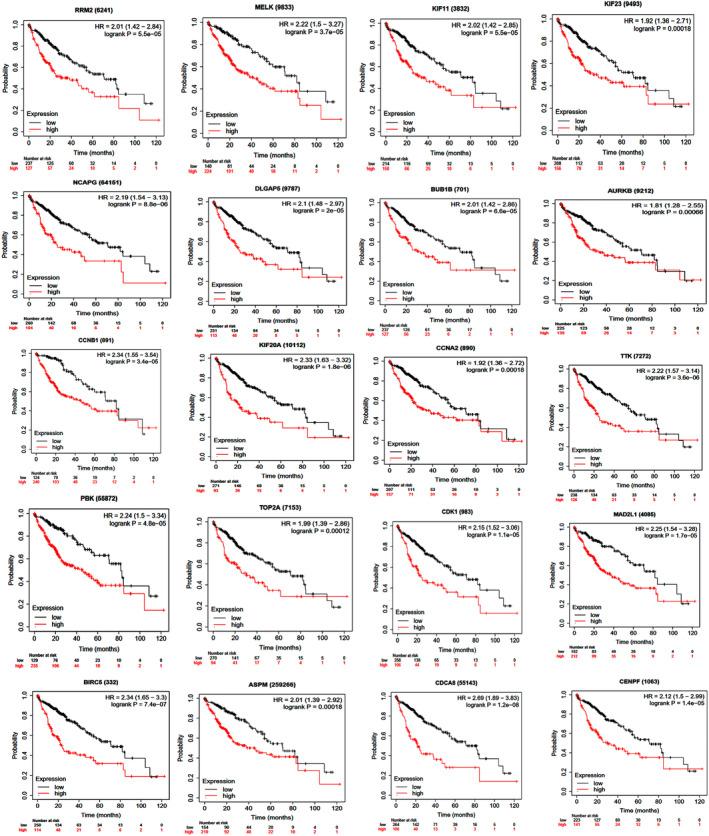
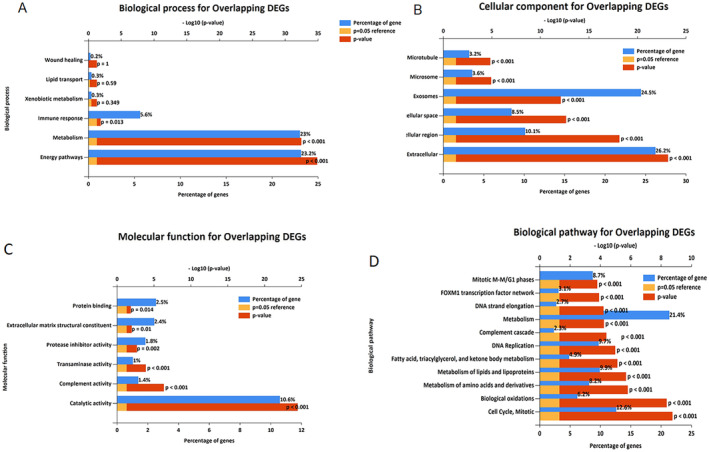
Common DEGs were identified, and a PPI network was constructed using the STRING database and Cytoscape software to identify hub genes. The reduced levels of tumor suppressor miRNAs or down regulated DEmiRs may be increased levels of oncogenes, the oncomirs or up regulated DEmiRs may be decreased levels of tumor suppressor genes in cancerous cells. According to this strategy, increased and decreased DEGs, also increased and decreased DEmiRs were selected. The multimir package was employed to predict target genes for DEmiRs then DEmiRs-hub gene network created. We identified approximately 1000 overlapping DEGs and 60 DEmiRs. Hub genes included RRM2, MELK, KIF11, KIF23, NCAPG, DLGAP5, BUB1B, AURKB, CCNB1, KIF20A, CCNA2, TTK, PBK, TOP2A, CDK1, MAD2L1, BIRC5, ASPM, CDCA8, and CENPF, all associated with significantly worse survival in HCC. miR-224, miR-24, miR-182, miRNA-1-3p, miR-30a, miR-27a, and miR-214 were identified as important DEmiRs with targeting more than six hub genes.
Generally, our findings offer insight into the interaction of hub genes and miRNAs in the development of HCC by bioinformatics analysis, information that may prove useful in identifying biomarkers and therapeutic targets in HCC.
肝细胞癌(HCC)表达谱的生物信息学分析有助于理解其分子机制,并确定新的诊断和治疗靶点。
在本研究中,我们使用R软件分析了来自基因表达综合数据库(GEO)的与HCC相关的表达谱数据集和miRNA表达谱,以检测差异表达基因(DEGs)和差异表达miRNAs(DEmiRs)。
鉴定出常见的DEGs,并使用STRING数据库和Cytoscape软件构建蛋白质-蛋白质相互作用(PPI)网络以识别枢纽基因。肿瘤抑制miRNAs水平降低或下调的DEmiRs可能是癌基因水平升高,癌miRNAs或上调的DEmiRs可能是癌细胞中肿瘤抑制基因水平降低。根据这一策略,选择了上调和下调的DEGs,以及上调和下调的DEmiRs。使用multimir软件包预测DEmiRs的靶基因,然后创建DEmiRs-枢纽基因网络。我们鉴定出约1000个重叠的DEGs和60个DEmiRs。枢纽基因包括RRM2、MELK、KIF11、KIF23、NCAPG、DLGAP5、BUB1B、AURKB、CCNB1、KIF20A、CCNA2、TTK、PBK、TOP2A、CDK1、MAD2L1、BIRC5、ASPM、CDCA8和CENPF,所有这些基因都与HCC患者显著较差的生存率相关。miR-224、miR-24、miR-182、miRNA-1-3p、miR-30a、miR-27a和miR-214被鉴定为重要的DEmiRs,它们靶向六个以上的枢纽基因。
总体而言,我们的研究结果通过生物信息学分析深入了解了枢纽基因和miRNAs在HCC发生发展中的相互作用,这些信息可能有助于识别HCC中的生物标志物和治疗靶点。