Yang Boxuan, Yang Jianbo, Chen Routing, Chai Jianmin, Wei Xiaoyuan, Zhao Jiangchao, Zhao Yunxiang, Deng Feilong, Li Ying
Guangdong Provincial Key Laboratory of Animal Molecular Design and Precise Breeding, Foshan University, Foshan 528225, China.
School of Animal Science and Technology, Foshan University, Foshan 528225, China.
Microorganisms. 2024 Nov 24;12(12):2409. doi: 10.3390/microorganisms12122409.
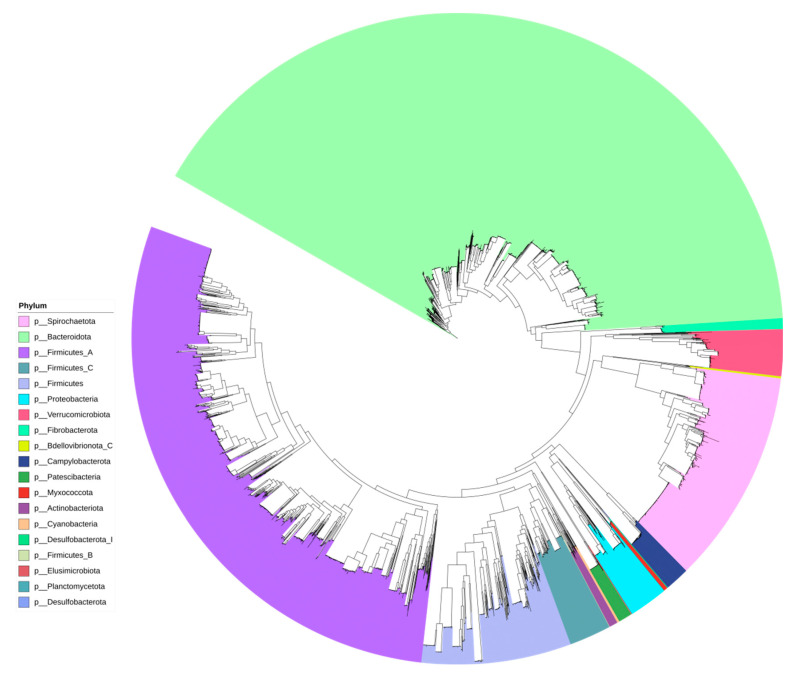
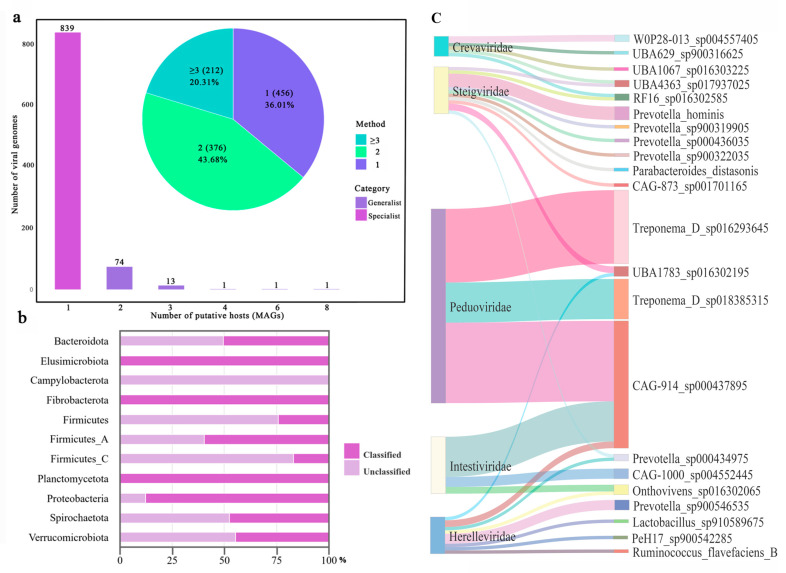
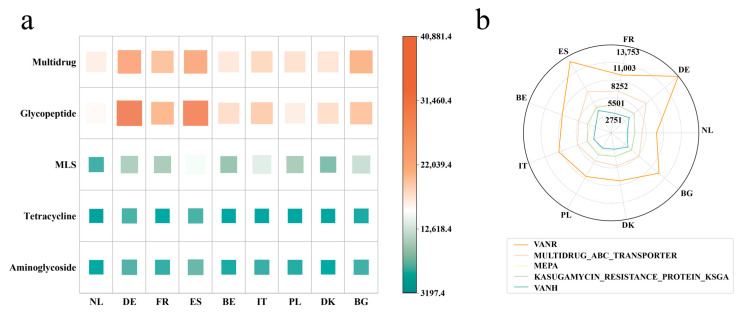
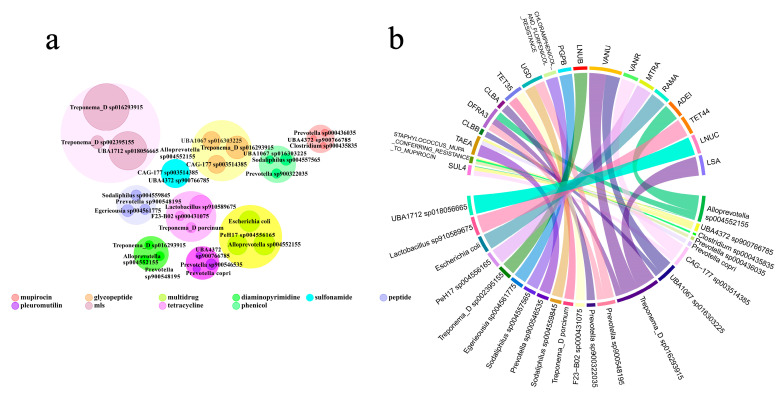
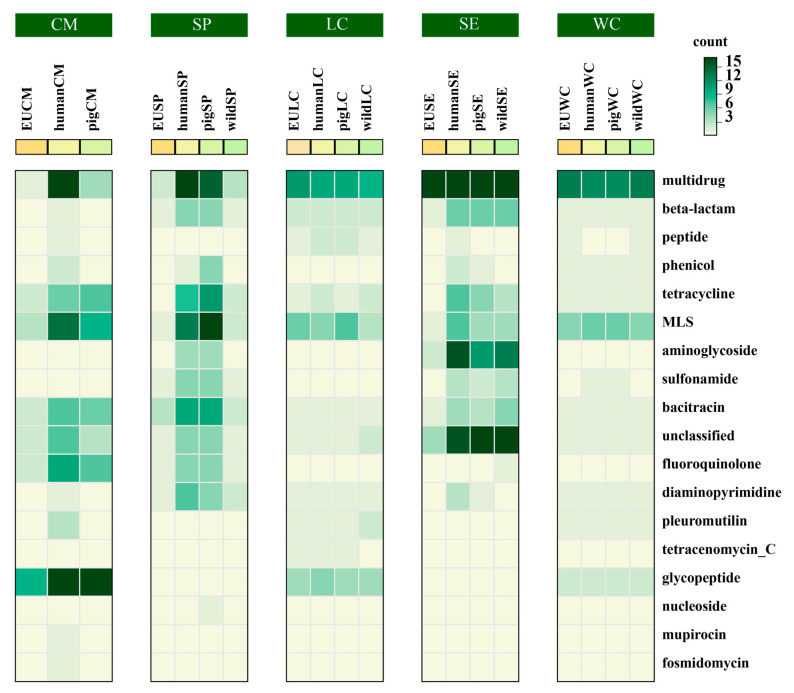
Gut microbiota plays a crucial role in the health and productivity of pigs. However, the spread of antibiotic resistance genes (ARGs) and viruses within the pig intestinal microbiota poses significant threats to animal and public health. This study utilized 181 pig samples from nine European countries and employed metagenomic assembly methods to investigate the dynamics and distribution of ARGs and viruses within the pig intestinal microbiota, aiming to observing their associations with potential bacterial hosts. We identified 4605 metagenome-assembled genomes (MAGs), corresponding to 19 bacterial phyla, 97 families, 309 genera, and a total of 449 species. Additionally, 44 MAGs were classified as archaea. Analysis of ARGs revealed 276 ARG types across 21 ARG classes, with Glycopeptide being the most abundant ARG class, followed by the class of Multidrug. was identified as a primary potential bacterial host for Glycopeptide. Aligning nucleotide sequences with a viral database, we identified 1044 viruses. Among the viral genome families, Peduoviridae and Intestiviridae were the most prevalent, with being the most common potential host species for both. These findings highlight the importance of MAGs in enhancing our understanding of the gut microbiome, revealing microbial diversity, antibiotic resistance, and virus-bacteria interactions. The data analysis for the article was based on the public dataset PRJEB22062 in the European Nucleotide Archive.
肠道微生物群对猪的健康和生产性能起着至关重要的作用。然而,抗生素抗性基因(ARGs)和病毒在猪肠道微生物群中的传播对动物和公共卫生构成了重大威胁。本研究利用来自9个欧洲国家的181份猪样本,采用宏基因组组装方法研究ARGs和病毒在猪肠道微生物群中的动态变化和分布,旨在观察它们与潜在细菌宿主的关联。我们鉴定出4605个宏基因组组装基因组(MAGs),对应19个细菌门、97个科、309个属,共449个物种。此外,44个MAGs被归类为古细菌。对ARGs的分析揭示了21种ARG类别中的276种ARG类型,其中糖肽类是最丰富的ARG类别,其次是多药类。 被确定为糖肽类的主要潜在细菌宿主。将核苷酸序列与病毒数据库进行比对,我们鉴定出1044种病毒。在病毒基因组家族中,短尾病毒科和肠道病毒科最为普遍, 是这两者最常见的潜在宿主物种。这些发现凸显了MAGs在增进我们对肠道微生物群的理解、揭示微生物多样性、抗生素抗性以及病毒 - 细菌相互作用方面的重要性。本文的数据分析基于欧洲核苷酸档案馆中的公共数据集PRJEB22062。