Kunkel Deborah, Sørensen Peter, Shankar Vijay, Morgante Fabio
School of Mathematical and Statistical Sciences, Clemson University, Clemson, South Carolina, United States of America.
Center for Quantitative Genetics and Genomics, Aarhus University, Aarhus, Denmark.
PLoS Genet. 2025 Jan 7;21(1):e1011519. doi: 10.1371/journal.pgen.1011519. eCollection 2025 Jan.
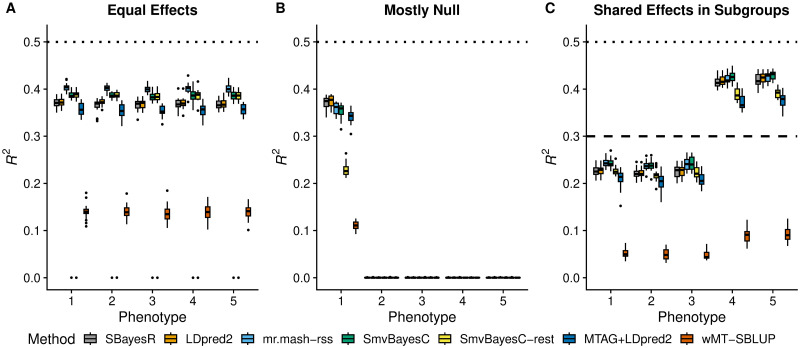
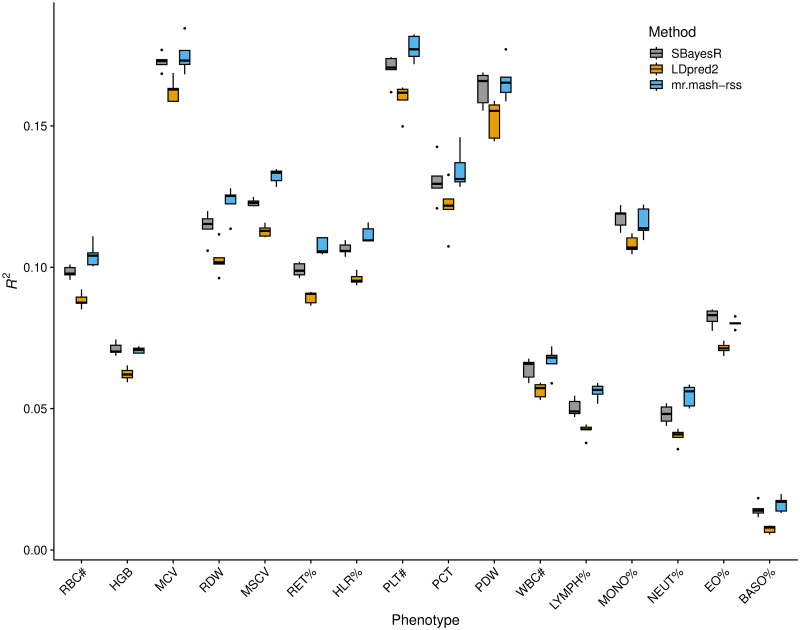
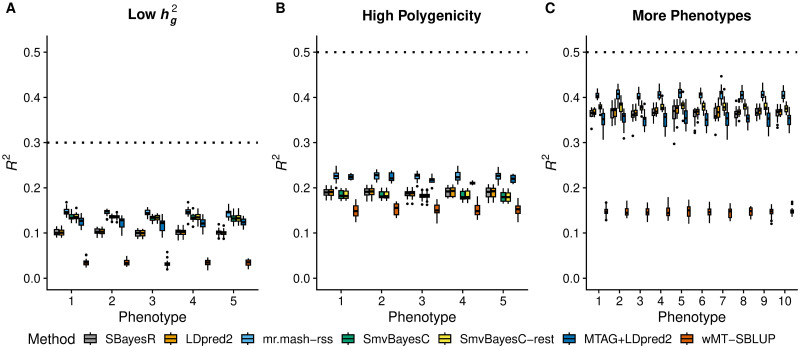
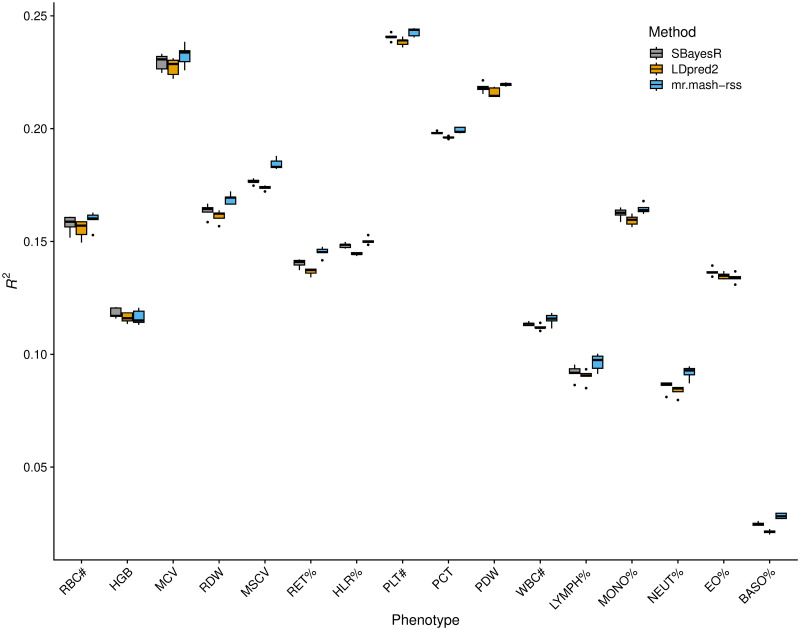
Polygenic prediction of complex trait phenotypes has become important in human genetics, especially in the context of precision medicine. Recently, mr.mash, a flexible and computationally efficient method that models multiple phenotypes jointly and leverages sharing of effects across such phenotypes to improve prediction accuracy, was introduced. However, a drawback of mr.mash is that it requires individual-level data, which are often not publicly available. In this work, we introduce mr.mash-rss, an extension of the mr.mash model that requires only summary statistics from Genome-Wide Association Studies (GWAS) and linkage disequilibrium (LD) estimates from a reference panel. By using summary data, we achieve the twin goal of increasing the applicability of the mr.mash model to data sets that are not publicly available and making it scalable to biobank-size data. Through simulations, we show that mr.mash-rss is competitive with, and often outperforms, current state-of-the-art methods for single- and multi-phenotype polygenic prediction in a variety of scenarios that differ in the pattern of effect sharing across phenotypes, the number of phenotypes, the number of causal variants, and the genomic heritability. We also present a real data analysis of 16 blood cell phenotypes in the UK Biobank, showing that mr.mash-rss achieves higher prediction accuracy than competing methods for the majority of traits, especially when the data set has smaller sample size.
复杂性状表型的多基因预测在人类遗传学中变得越来越重要,尤其是在精准医学的背景下。最近,mr.mash被引入,这是一种灵活且计算高效的方法,它联合对多个表型进行建模,并利用这些表型间效应的共享来提高预测准确性。然而,mr.mash的一个缺点是它需要个体水平的数据,而这些数据通常并非公开可用。在这项工作中,我们引入了mr.mash-rss,它是mr.mash模型的扩展,仅需要全基因组关联研究(GWAS)的汇总统计数据以及来自参考面板的连锁不平衡(LD)估计值。通过使用汇总数据,我们实现了双重目标:提高mr.mash模型对非公开可用数据集的适用性,并使其能够扩展到生物样本库规模的数据。通过模拟,我们表明在表型间效应共享模式、表型数量、因果变异数量和基因组遗传力不同的各种场景中,mr.mash-rss在单表型和多表型多基因预测方面与当前最先进的方法具有竞争力,并且常常表现更优。我们还对英国生物样本库中的16种血细胞表型进行了实际数据分析,结果表明对于大多数性状,mr.mash-rss比竞争方法具有更高的预测准确性,尤其是当数据集样本量较小时。