Fortuno Cristina, Llinares-Burguet Inés, Canson Daffodil M, de la Hoya Miguel, Bueno-Martínez Elena, Sanoguera-Miralles Lara, Caldes Sonsoles, James Paul A, Velasco-Sampedro Eladio A, Spurdle Amanda B
Population Health Program, QIMR Berghofer Medical Research Institute, Herston, QLD, 4006, Australia.
Splicing and Genetic Susceptibility to Cancer, Instituto de Biomedicina y Genética Molecular de Valladolid (IBGM), Consejo Superior de Investigaciones Científicas - Universidad de Valladolid (CSIC-UVa), 47003, Valladolid, Spain.
Hum Genomics. 2025 Jan 8;19(1):2. doi: 10.1186/s40246-024-00714-5.
TP53 variant classification benefits from the availability of large-scale functional data for missense variants generated using cDNA-based assays. However, absence of comprehensive splicing assay data for TP53 confounds the classification of the subset of predicted missense and synonymous variants that are also predicted to alter splicing. Our study aimed to generate and apply splicing assay data for a prioritised group of 59 TP53 predicted missense or synonymous variants that are also predicted to affect splicing by either SpliceAI or MaxEntScan.
We conducted splicing analyses using a minigene construct containing TP53 exons 2 to 9 transfected into human breast cancer SKBR3 cells, and compared results against different splice prediction methods, including correlation with the SpliceAI-10k calculator. We additionally applied the splicing results for TP53 variant classification using an approach consistent with the ClinGen Sequence Variant Interpretation Splicing Subgroup recommendations.
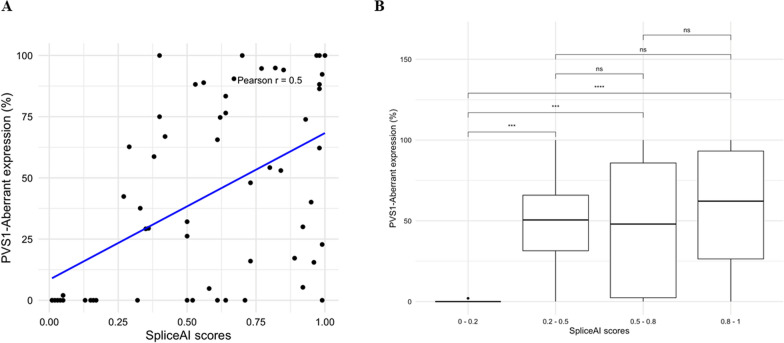
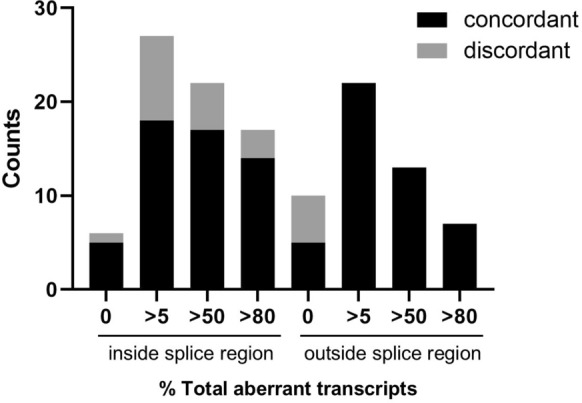
Aberrant transcript profile consistent with loss of function, and for which a PVS1 (RNA) code would be assigned, was observed for 42 (71%) of prioritised variants, of which aberrant transcript expression was over 50% for 26 variants, and over 80% for 15 variants. Data supported the use of SpliceAI ≥ 0.2 cutoff for predicted splicing impact of TP53 variants. Prediction of aberration types using SpliceAI-10k calculator generally aligned with the corresponding assay results, though maximum SpliceAI score did not accurately predict level of aberrant expression. Application of the observed splicing results was used to reclassify 27/59 (46%) test variants as (likely) pathogenic or (likely) benign.
In conclusion, this study enhances the integration of splicing predictions and provides splicing assay data for exonic variants to support TP53 germline classification.
TP53变异分类受益于使用基于cDNA的检测方法生成的错义变异大规模功能数据。然而,缺乏TP53的全面剪接检测数据使得预测的错义变异和同义变异子集中那些也被预测会改变剪接的变异的分类变得复杂。我们的研究旨在为一组经SpliceAI或MaxEntScan预测也会影响剪接的59个TP53预测错义或同义变异生成并应用剪接检测数据。
我们使用一个包含TP53外显子2至9的小基因构建体转染到人乳腺癌SKBR3细胞中进行剪接分析,并将结果与不同的剪接预测方法进行比较,包括与SpliceAI-10k计算器的相关性。我们还使用与ClinGen序列变异解释剪接亚组建议一致的方法将TP53变异分类的剪接结果进行应用。
在42个(71%)优先变异中观察到与功能丧失一致的异常转录本谱,对于这些变异将指定PVS1(RNA)编码,其中26个变异的异常转录本表达超过50%,15个变异超过80%。数据支持使用SpliceAI≥0.2的截断值来预测TP53变异的剪接影响。使用SpliceAI-10k计算器预测的畸变类型通常与相应的检测结果一致,尽管最大SpliceAI分数不能准确预测异常表达水平。观察到的剪接结果的应用用于将27/59(46%)的测试变异重新分类为(可能)致病或(可能)良性。
总之,本研究加强了剪接预测的整合,并为外显子变异提供了剪接检测数据以支持TP53种系分类。