Elzinga Sarah E, Guo Kai, Turfah Ali, Henn Rosemary E, Webber-Davis Ian F, Hayes John M, Pacut Crystal M, Teener Samuel J, Carter Andrew D, Rigan Diana M, Allouch Adam M, Jang Dae-Gyu, Parent Rachel, Glass Emily, Murphy Geoffrey G, Lentz Stephen I, Chen Kevin S, Zhao Lili, Hur Junguk, Feldman Eva L
Department of Neurology, University of Michigan, Ann Arbor, Michigan, USA.
Department of Physiology, Michigan State University, East Lansing, Michigan, USA.
Alzheimers Dement. 2025 Mar;21(3):e70060. doi: 10.1002/alz.70060.
Metabolic stressors (obesity, metabolic syndrome, prediabetes, and type 2 diabetes [T2D]) increase the risk of cognitive impairment (CI), including Alzheimer's disease (AD). Immune system dysregulation and inflammation, particularly microglial mediated, may underlie this risk, but mechanisms remain unclear.
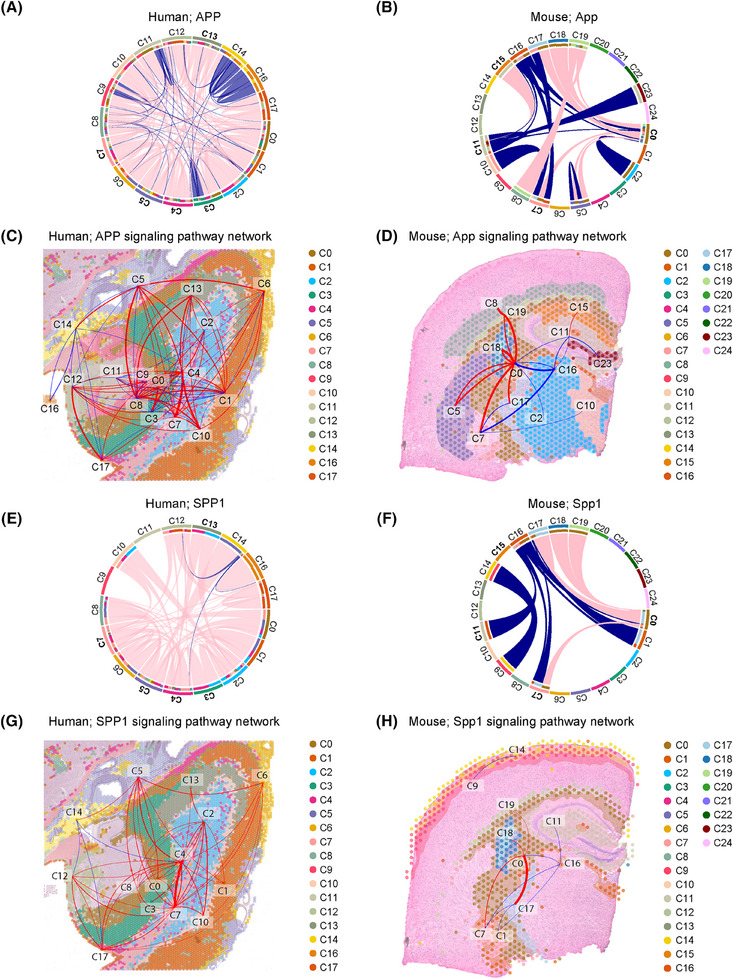
Using a high-fat diet-fed (HFD) model, we assessed longitudinal metabolism and cognition, and terminal inflammation and brain spatial transcriptomics. Additionally, we performed hippocampal spatial transcriptomics and single-cell RNA sequencing of post mortem tissue from AD and T2D human subjects versus controls.
HFD induced progressive metabolic and CI with terminal inflammatory changes, and dysmetabolic, neurodegenerative, and inflammatory gene expression profiles, particularly in microglia. AD and T2D human subjects had similar gene expression changes, including in secreted phosphoprotein 1 (SPP1), a pro-inflammatory gene associated with AD.
These data show that metabolic stressors cause early and progressive CI, with inflammatory changes that promote disease. They also indicate a role for microglia, particularly microglial SPP1, in CI.
Metabolic stress causes persistent metabolic and cognitive impairments in mice. Murine and human brain spatial transcriptomics align and indicate a pro-inflammatory milieu. Transcriptomic data indicate a role for microglial-mediated inflammatory mechanisms. Secreted phosphoprotein 1 emerged as a potential target of interest in metabolically driven cognitive impairment.
代谢应激源(肥胖、代谢综合征、糖尿病前期和2型糖尿病[T2D])会增加认知障碍(CI)的风险,包括阿尔茨海默病(AD)。免疫系统失调和炎症,尤其是小胶质细胞介导的炎症,可能是这种风险的基础,但具体机制仍不清楚。
我们使用高脂饮食喂养(HFD)模型,评估了纵向代谢和认知,以及终末期炎症和脑空间转录组学。此外,我们对AD和T2D人类受试者与对照组的死后组织进行了海马空间转录组学和单细胞RNA测序。
HFD诱导了渐进性代谢和CI,并伴有终末期炎症变化,以及代谢异常、神经退行性变和炎症基因表达谱,特别是在小胶质细胞中。AD和T2D人类受试者有类似的基因表达变化,包括在分泌磷蛋白1(SPP1)中,这是一种与AD相关的促炎基因。
这些数据表明,代谢应激源会导致早期和渐进性CI,并伴有促进疾病的炎症变化。它们还表明小胶质细胞,特别是小胶质细胞SPP1在CI中发挥作用。
代谢应激会导致小鼠持续的代谢和认知障碍。小鼠和人类脑空间转录组学结果一致,并表明存在促炎环境。转录组学数据表明小胶质细胞介导的炎症机制发挥了作用。分泌磷蛋白1成为代谢驱动的认知障碍中一个潜在的重要靶点。