Pelto Juho, Auranen Kari, Kujala Janne V, Lahti Leo
Department of Computing, University of Turku, University of Turku, 20014, Finland.
Department of Mathematics and Statistics, University of Turku, University of Turku, 20014, Finland.
Brief Bioinform. 2025 Mar 4;26(2). doi: 10.1093/bib/bbaf130.
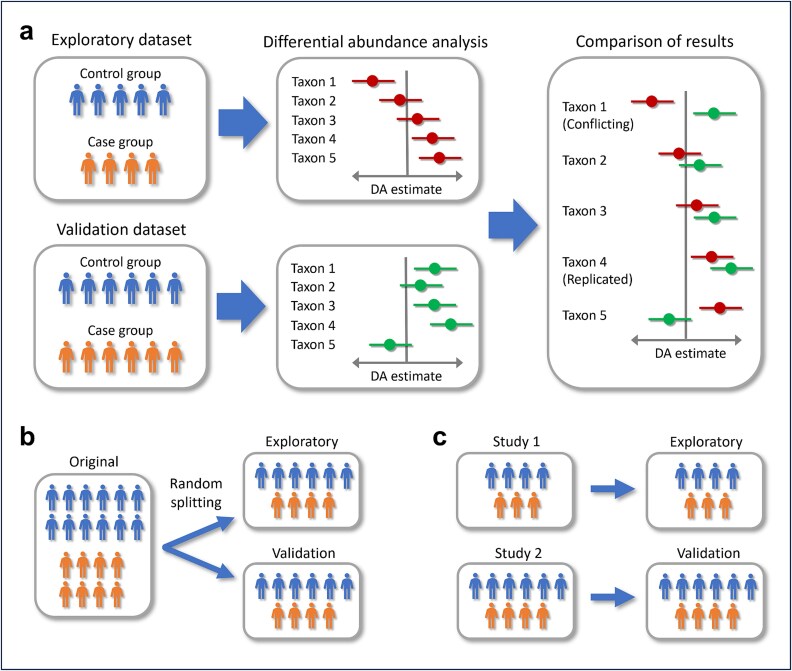
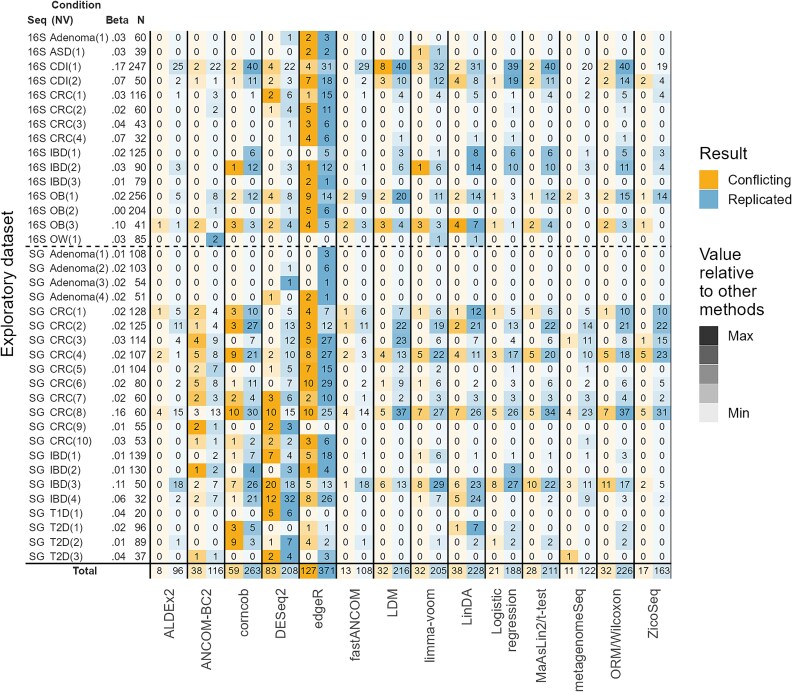
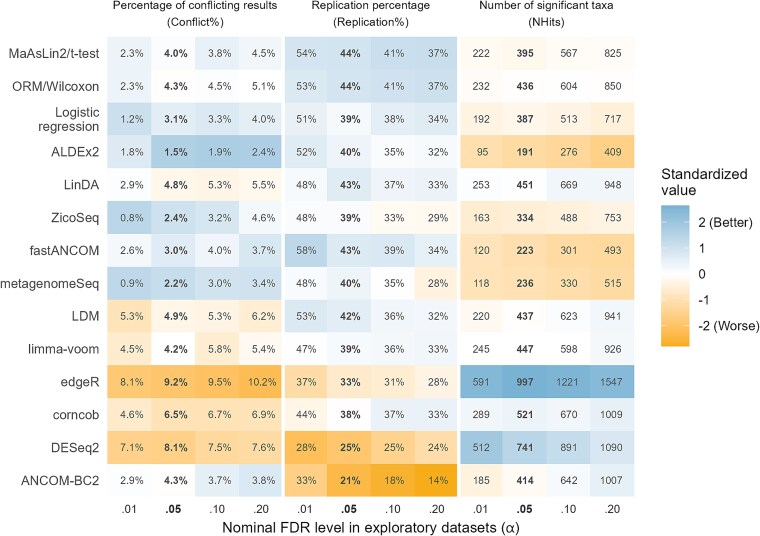
Differential abundance analysis (DAA) is a key component of microbiome studies. Although dozens of methods exist, there is currently no consensus on the preferred methods. While the correctness of results in DAA is an ambiguous concept and cannot be fully evaluated without setting the ground truth and employing simulated data, we argue that a well-performing method should be effective in producing highly reproducible results. We compared the performance of 14 DAA methods by employing datasets from 53 taxonomic profiling studies based on 16S rRNA gene or shotgun metagenomic sequencing. For each method, we examined how the results replicated between random partitions of each dataset and between datasets from separate studies. While certain methods showed good consistency, some widely used methods were observed to produce a substantial number of conflicting findings. Overall, when considering consistency together with sensitivity, the best performance was attained by analyzing relative abundances with a nonparametric method (Wilcoxon test or ordinal regression model) or linear regression/t-test. Moreover, a comparable performance was obtained by analyzing presence/absence of taxa with logistic regression.
差异丰度分析(DAA)是微生物组研究的关键组成部分。尽管存在数十种方法,但目前对于首选方法尚无共识。虽然DAA结果的正确性是一个模糊的概念,在没有设定基本事实和使用模拟数据的情况下无法完全评估,但我们认为一种性能良好的方法应该能够有效地产生高度可重复的结果。我们通过使用来自53项基于16S rRNA基因或鸟枪法宏基因组测序的分类学分析研究的数据集,比较了14种DAA方法的性能。对于每种方法,我们研究了结果在每个数据集的随机划分之间以及来自不同研究的数据集之间的复制情况。虽然某些方法显示出良好的一致性,但观察到一些广泛使用的方法产生了大量相互矛盾的结果。总体而言,在考虑一致性和敏感性时,通过使用非参数方法(Wilcoxon检验或有序回归模型)或线性回归/t检验分析相对丰度可获得最佳性能。此外,通过使用逻辑回归分析分类单元的存在/不存在也可获得类似的性能。