Department of Agricultural Sciences, University of Naples Federico II, Portici, Italy.
Task Force on Microbiome Studies, University of Naples Federico II, Naples, Italy.
PLoS Comput Biol. 2022 Apr 21;18(4):e1010066. doi: 10.1371/journal.pcbi.1010066. eCollection 2022 Apr.
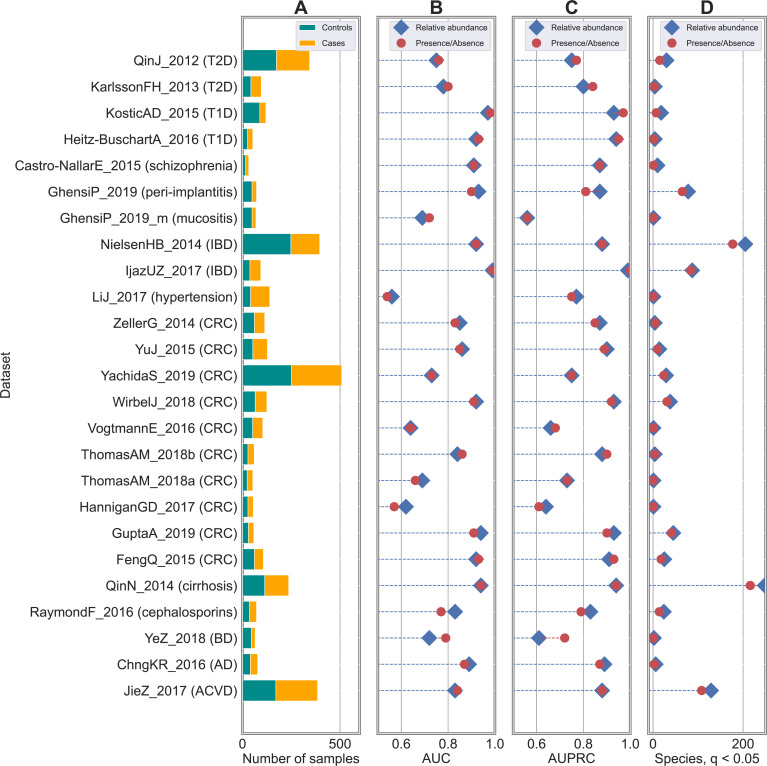
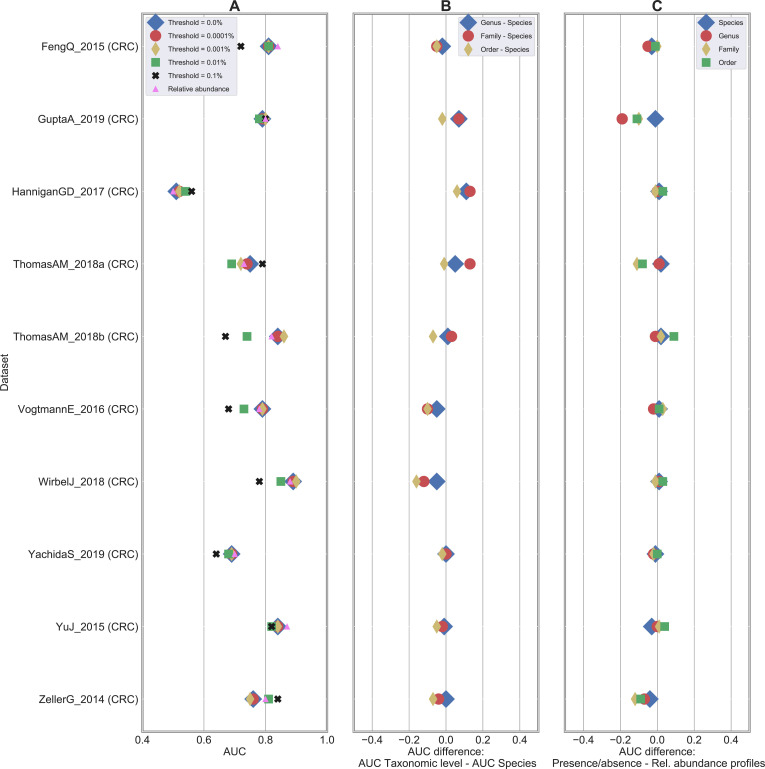
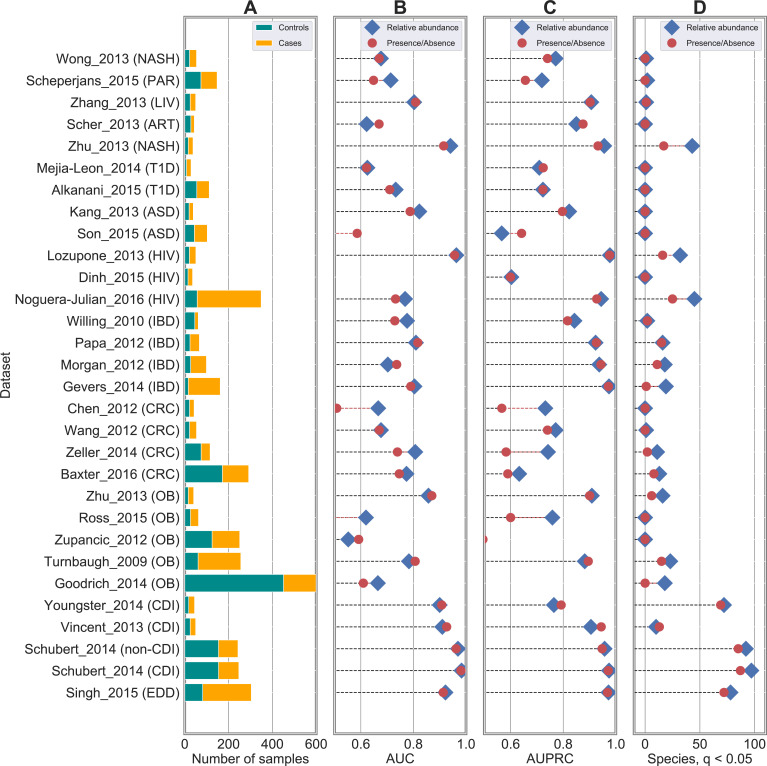
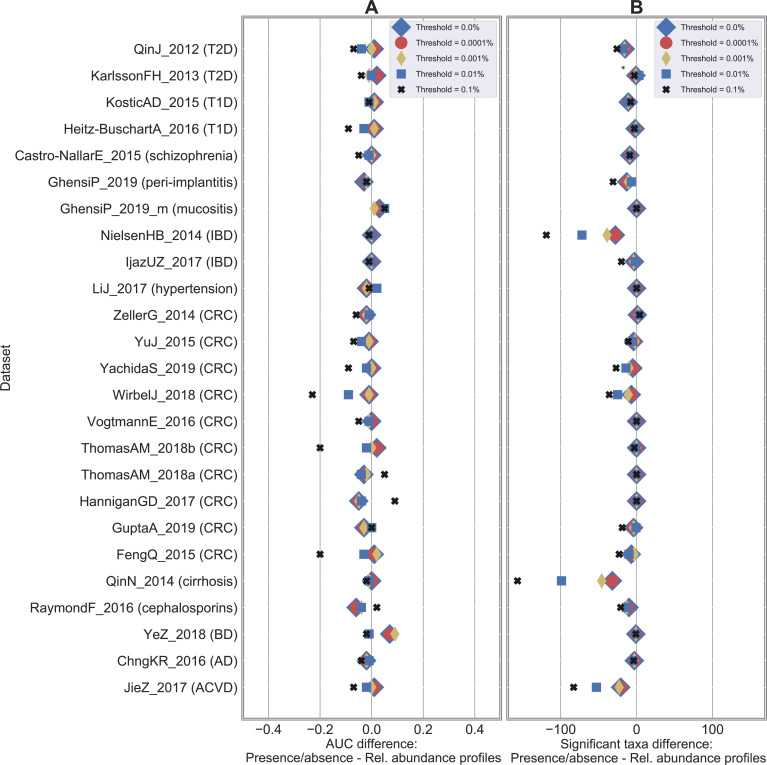
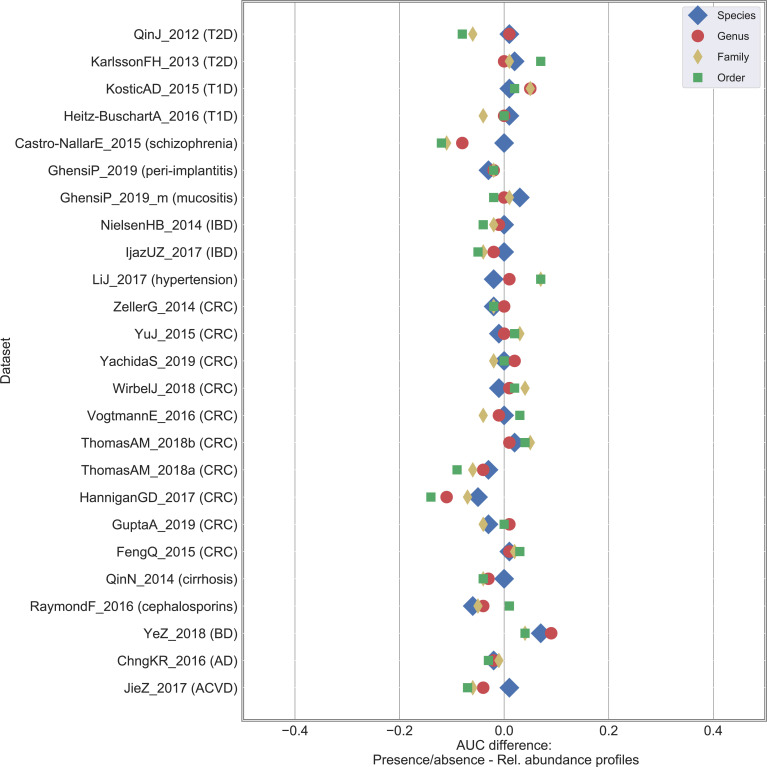
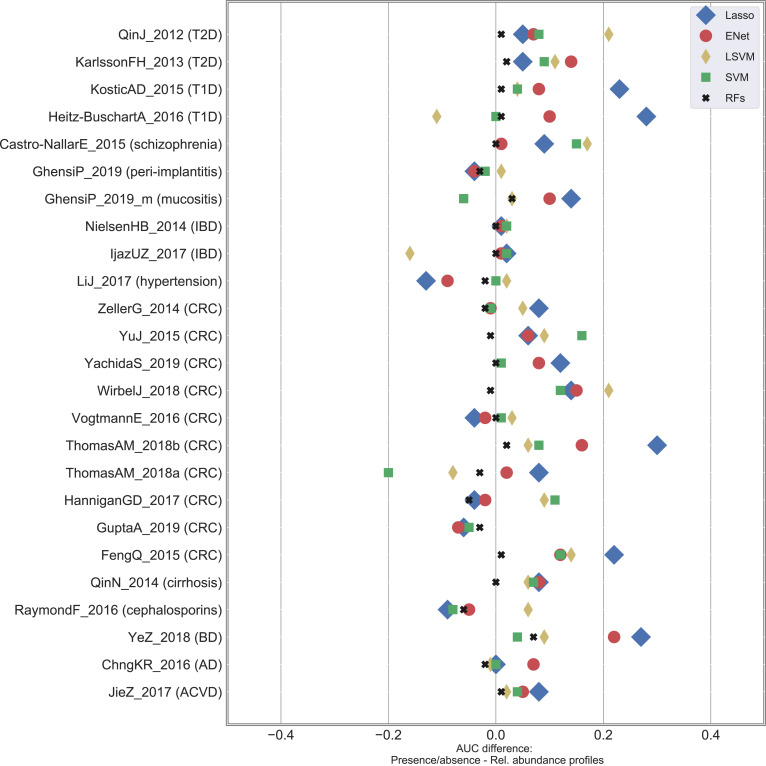
Machine learning-based classification approaches are widely used to predict host phenotypes from microbiome data. Classifiers are typically employed by considering operational taxonomic units or relative abundance profiles as input features. Such types of data are intrinsically sparse, which opens the opportunity to make predictions from the presence/absence rather than the relative abundance of microbial taxa. This also poses the question whether it is the presence rather than the abundance of particular taxa to be relevant for discrimination purposes, an aspect that has been so far overlooked in the literature. In this paper, we aim at filling this gap by performing a meta-analysis on 4,128 publicly available metagenomes associated with multiple case-control studies. At species-level taxonomic resolution, we show that it is the presence rather than the relative abundance of specific microbial taxa to be important when building classification models. Such findings are robust to the choice of the classifier and confirmed by statistical tests applied to identifying differentially abundant/present taxa. Results are further confirmed at coarser taxonomic resolutions and validated on 4,026 additional 16S rRNA samples coming from 30 public case-control studies.
基于机器学习的分类方法被广泛用于从微生物组数据中预测宿主表型。分类器通常通过考虑操作分类单元或相对丰度谱作为输入特征来使用。这种类型的数据本质上是稀疏的,这为从微生物分类群的存在/不存在而不是相对丰度进行预测提供了机会。这也提出了一个问题,即对于区分目的,是否是特定分类群的存在而不是丰度相关,这一方面在文献中迄今为止被忽视了。在本文中,我们旨在通过对 4128 个与多个病例对照研究相关的公开宏基因组进行荟萃分析来填补这一空白。在物种水平的分类分辨率下,我们表明,在构建分类模型时,重要的是特定微生物分类群的存在而不是相对丰度。这些发现不受分类器选择的影响,并通过应用于识别差异丰富/存在的分类群的统计检验得到证实。结果在更粗的分类分辨率下得到进一步证实,并在来自 30 个公共病例对照研究的另外 4026 个 16S rRNA 样本上得到验证。