Shamash Michael, Sinha Anshul, Maurice Corinne F
Department of Microbiology & Immunology, McGill University, Montreal, Quebec, Canada.
McGill Centre for Microbiome Research, Montreal, Quebec, Canada.
mSystems. 2025 May 20;10(5):e0136424. doi: 10.1128/msystems.01364-24. Epub 2025 Apr 8.
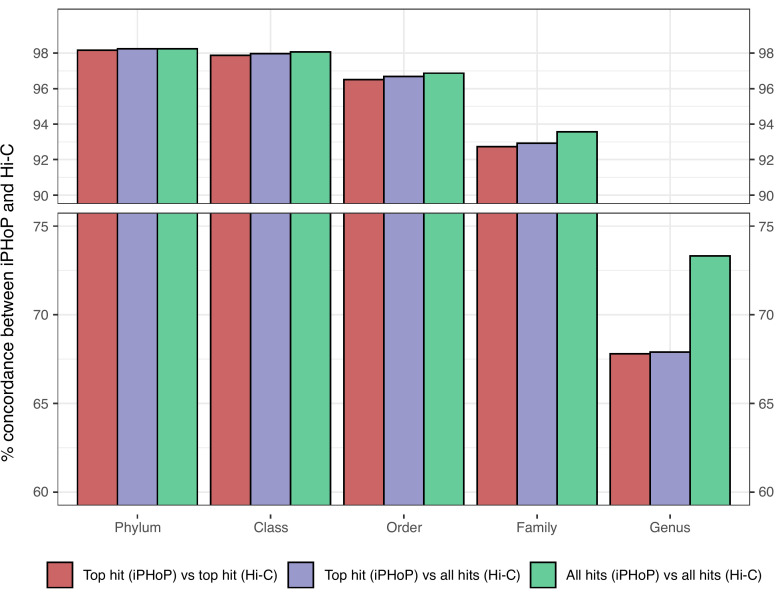
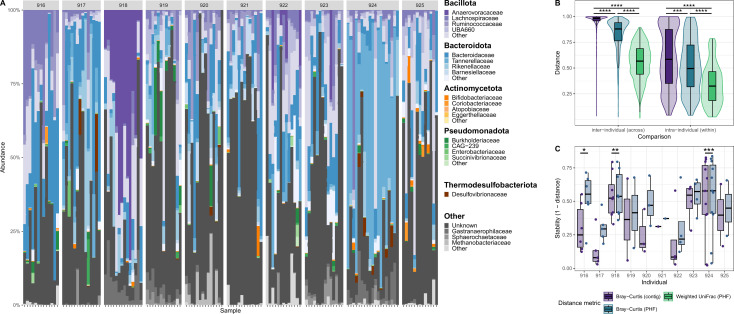
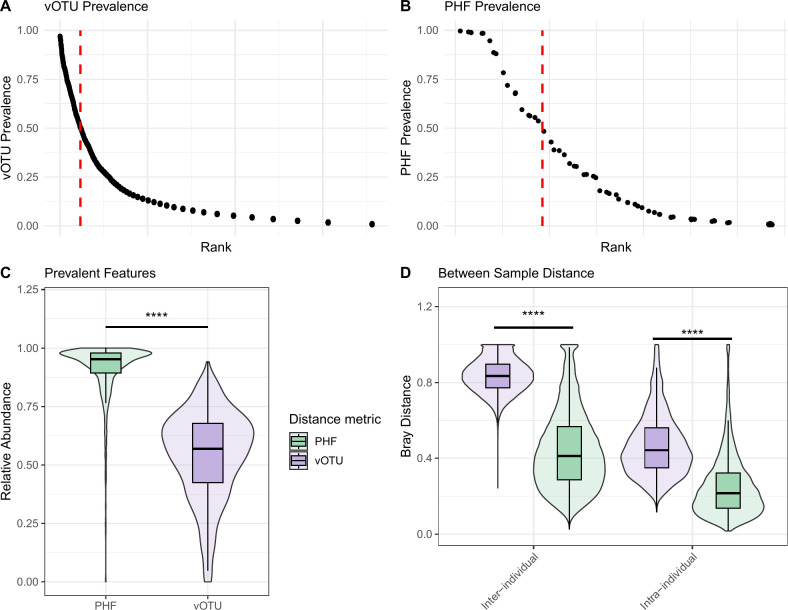
The human gut virome is predominantly made up of bacteriophages (phages), viruses that infect bacteria. Metagenomic studies have revealed that phages in the gut are highly individual specific and dynamic. These features make it challenging to perform meaningful cross-study comparisons. While several taxonomy frameworks exist to group phages and improve these comparisons, these strategies provide little insight into the potential effects phages have on their bacterial hosts. Here, we propose the use of predicted phage host families (PHFs) as a functionally relevant, qualitative unit of phage classification to improve these cross-study analyses. We first show that bioinformatic predictions of phage hosts are accurate at the host family level by measuring their concordance to Hi-C sequencing-based predictions in human and mouse fecal samples. Next, using phage host family predictions, we determined that PHFs reduce intra- and interindividual ecological distances compared to viral contigs in a previously published cohort of 10 healthy individuals, while simultaneously improving longitudinal virome stability. Lastly, by reanalyzing a previously published metagenomics data set with >1,000 samples, we determined that PHFs are prevalent across individuals and can aid in the detection of inflammatory bowel disease-specific virome signatures. Overall, our analyses support the use of predicted phage hosts in reducing between-sample distances and providing a biologically relevant framework for making between-sample virome comparisons.
The human gut virome consists mainly of bacteriophages (phages), which infect bacteria and show high individual specificity and variability, complicating cross-study comparisons. Furthermore, existing taxonomic frameworks offer limited insight into their interactions with bacterial hosts. In this study, we propose using predicted phage host families (PHFs) as a higher-level classification unit to enhance functional cross-study comparisons. We demonstrate that bioinformatic predictions of phage hosts align with Hi-C sequencing results at the host family level in human and mouse fecal samples. We further show that PHFs reduce ecological distances and improve virome stability over time. Additionally, reanalysis of a large metagenomics data set revealed that PHFs are widespread and can help identify disease-specific virome patterns, such as those linked to inflammatory bowel disease.
人类肠道病毒组主要由噬菌体(phages)组成,噬菌体是感染细菌的病毒。宏基因组学研究表明,肠道中的噬菌体具有高度个体特异性和动态性。这些特性使得进行有意义的跨研究比较具有挑战性。虽然存在几种分类框架来对噬菌体进行分组并改善这些比较,但这些策略对噬菌体对其细菌宿主的潜在影响提供的见解很少。在这里,我们建议使用预测的噬菌体宿主家族(PHFs)作为功能相关的噬菌体分类定性单位,以改善这些跨研究分析。我们首先通过测量人类和小鼠粪便样本中噬菌体宿主的生物信息学预测与基于Hi-C测序的预测的一致性,表明噬菌体宿主的生物信息学预测在宿主家族水平上是准确的。接下来,使用噬菌体宿主家族预测,我们确定在先前发表的一组10名健康个体中,与病毒重叠群相比,PHFs减少了个体内和个体间的生态距离,同时提高了纵向病毒组稳定性。最后,通过重新分析一个先前发表的包含1000多个样本的宏基因组学数据集,我们确定PHFs在个体中普遍存在,并有助于检测炎症性肠病特异性病毒组特征。总体而言,我们的分析支持使用预测的噬菌体宿主来减少样本间距离,并为进行样本间病毒组比较提供生物学相关框架。
人类肠道病毒组主要由噬菌体组成,噬菌体感染细菌并表现出高度个体特异性和变异性,使跨研究比较变得复杂。此外,现有的分类框架对它们与细菌宿主的相互作用提供的见解有限。在这项研究中,我们建议使用预测的噬菌体宿主家族(PHFs)作为更高层次的分类单位,以加强功能跨研究比较。我们证明,噬菌体宿主的生物信息学预测与人类和小鼠粪便样本中宿主家族水平的Hi-C测序结果一致。我们进一步表明,PHFs减少了生态距离,并随着时间的推移提高了病毒组稳定性。此外,对一个大型宏基因组学数据集的重新分析表明,PHFs广泛存在,并有助于识别疾病特异性病毒组模式,如与炎症性肠病相关的模式。