Elsayyid Malek, Tanis Jessica E, Yu Yanbao
Department of Biological Sciences, University of Delaware, Newark, Delaware 19716, United States.
Department of Chemistry and Biochemistry, University of Delaware, Newark, Delaware 19716, United States.
Anal Chem. 2025 May 6;97(17):9159-9167. doi: 10.1021/acs.analchem.4c05003. Epub 2025 Apr 21.
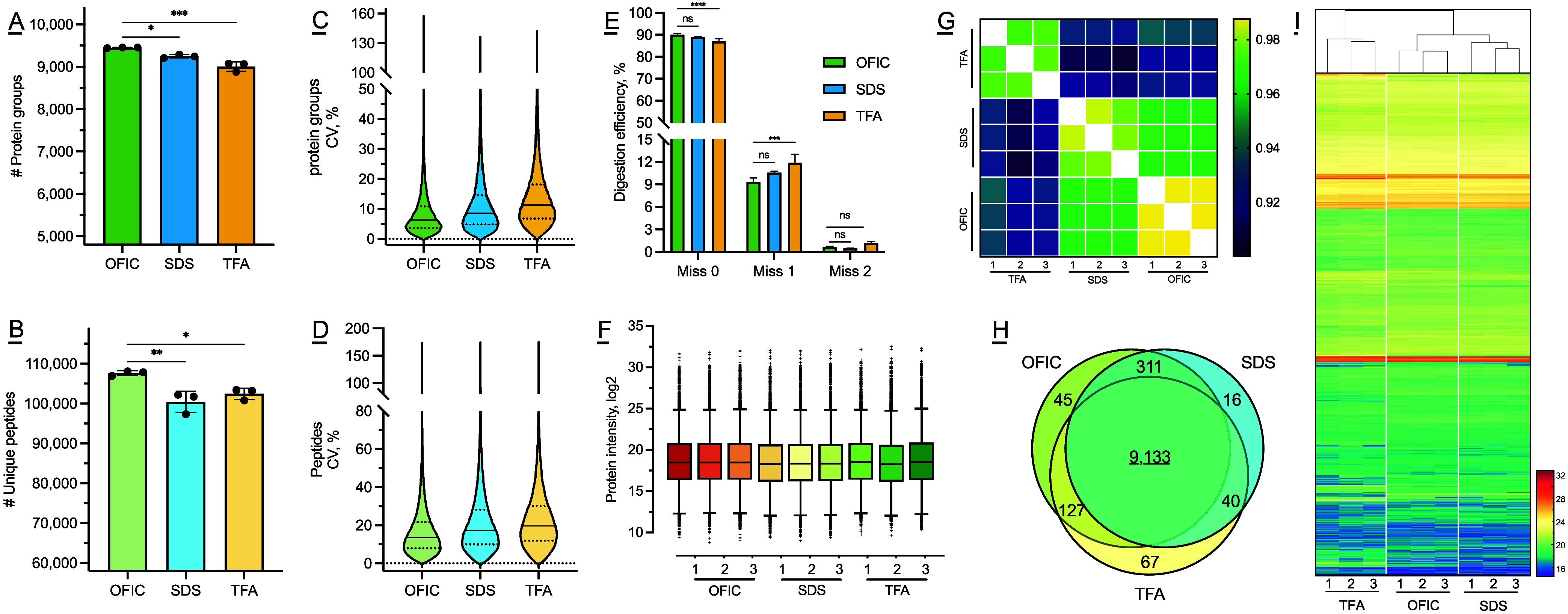
is a widely used genetic model organism; however, the worm cuticle complicates extraction of intracellular proteins, a prerequisite for typical bottom-up proteomics. Conventional physical disruption procedures are not only time-consuming but can also cause significant sample loss, making it difficult to perform proteomics with low-input samples. Here, for the first time, we present an on-filter in-cell (OFIC) processing approach that can digest proteins directly in the cells of the organism after methanol fixation. With OFIC processing and single-shot LC-MS analysis, we identified over 9400 proteins from a sample of only 200 worms, the largest proteome reported to date that did not require fractionation or enrichment. We systematically evaluated the performance of the OFIC approach by comparing it to conventional lysis-based methods. Our data suggest superior performance of OFIC processing for proteome identification and quantitation. We further evaluated the OFIC approach with even lower-input samples, including single worms. Then, we used this method to determine how the proteome is impacted by loss of superoxide dismutase , the ortholog of human , a gene associated with amyotrophic lateral sclerosis. Analysis of 8800 proteins from only 50 worms as the initial input showed that loss of affects the abundance of proteins required for stress response, ribosome biogenesis, and metabolism. In conclusion, our streamlined OFIC approach, which can be broadly applied to other systems, minimizes sample loss while offering the simplest workflow reported to date for proteomics.
是一种广泛使用的遗传模式生物;然而,线虫的角质层使细胞内蛋白质的提取变得复杂,而细胞内蛋白质提取是典型的自下而上蛋白质组学的一个先决条件。传统的物理破碎方法不仅耗时,还会导致大量样品损失,使得用低输入量样品进行蛋白质组学研究变得困难。在此,我们首次提出一种滤膜上细胞内(OFIC)处理方法,该方法可在甲醇固定后直接在生物体细胞内消化蛋白质。通过OFIC处理和单次液相色谱 - 质谱分析,我们从仅200条线虫的样品中鉴定出了超过9400种蛋白质,这是迄今为止报道的最大蛋白质组,且无需分级分离或富集。我们通过将OFIC方法与传统的基于裂解的方法进行比较,系统地评估了该方法的性能。我们的数据表明,OFIC处理在蛋白质组鉴定和定量方面具有卓越性能。我们进一步用甚至更低输入量的样品(包括单条线虫)评估了OFIC方法。然后,我们使用该方法来确定蛋白质组如何受到超氧化物歧化酶缺失的影响,超氧化物歧化酶是人类超氧化物歧化酶的直系同源物,该基因与肌萎缩侧索硬化症相关。以仅50条线虫作为初始输入对8800种蛋白质进行分析表明,超氧化物歧化酶的缺失会影响应激反应、核糖体生物发生和代谢所需蛋白质的丰度。总之,我们简化的OFIC方法可广泛应用于其他系统,在最大限度减少样品损失的同时,提供了迄今为止报道的用于线虫蛋白质组学最简单的工作流程。