Wang Shiguan, Han Pan, Mi Ping, Wang Chunxue, Lu Miao, Li Xinying, Xu Bowen, Wang Haoran, Gao Yingchen, Hou Yanlei, Tan Xueying, Liang Jinyuan, Ding Xue, Zhang Yan, Zhang Tingguo, Yuan Detian, Gao Lei, Zhang Cuijuan
Department of Clinical Laboratory, Qilu Hospital of Shandong University, Jinan, China; Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, China.
Department of Biochemistry and Molecular Biology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, China; State Key Laboratory of Cellular Stress Biology, School of Life Sciences, Xiamen University, Xiamen, China.
Cell Mol Gastroenterol Hepatol. 2025 Apr 24;19(9):101523. doi: 10.1016/j.jcmgh.2025.101523.
BACKGROUND & AIMS: Hepatocellular carcinoma (HCC) frequently involves metabolic reprogramming, which promotes oncogenesis and metastasis. However, the underlying molecular mechanisms remain insufficiently explored. In this study, we aim to investigate the metabolic abnormalities in c-Myc-driven HCC development and their potential therapeutic implications.
RNA sequencing and metabolomics were performed on HCC and adjacent tissues in a murine HCC model established by hydrodynamic tail-vein injection of c-Myc and sgTrp53/Cas9 plasmids. Key catalytic enzyme gene knockout was used to assess tumor formation and murine survival. Gene expression was analyzed using quantitative polymerase chain reaction, immunohistochemistry, and Western blot. Chromatin immunoprecipitation followed by quantitative polymerase chain reaction and luciferase assays verified c-Myc regulation.
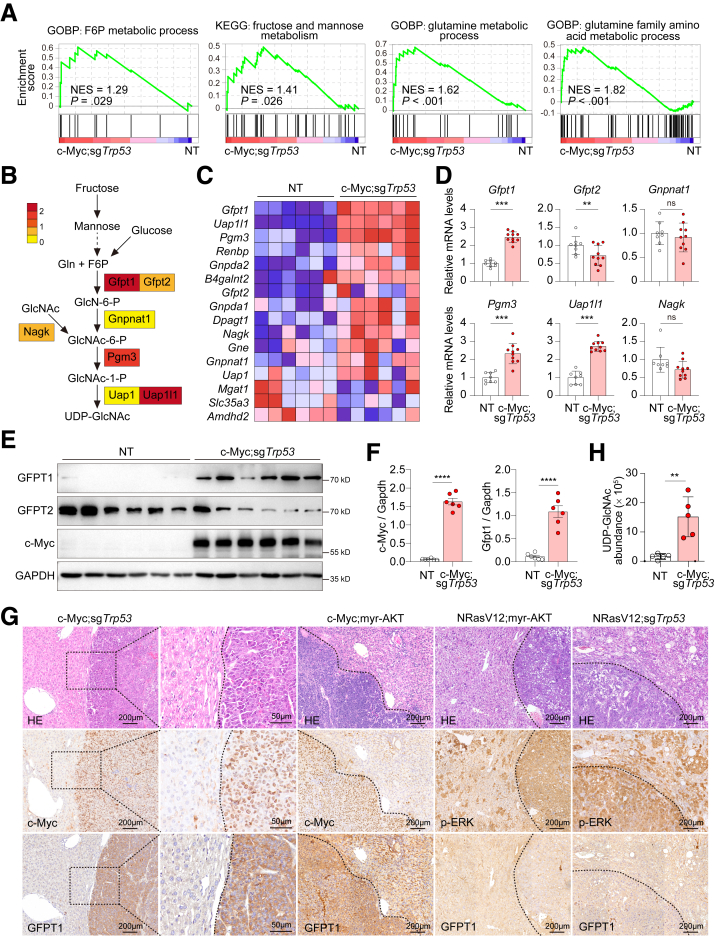
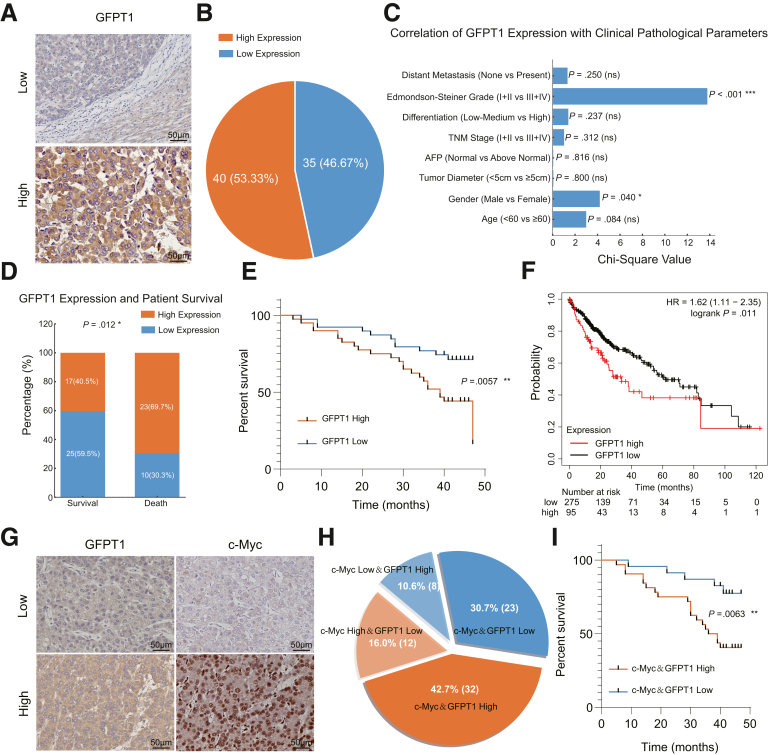
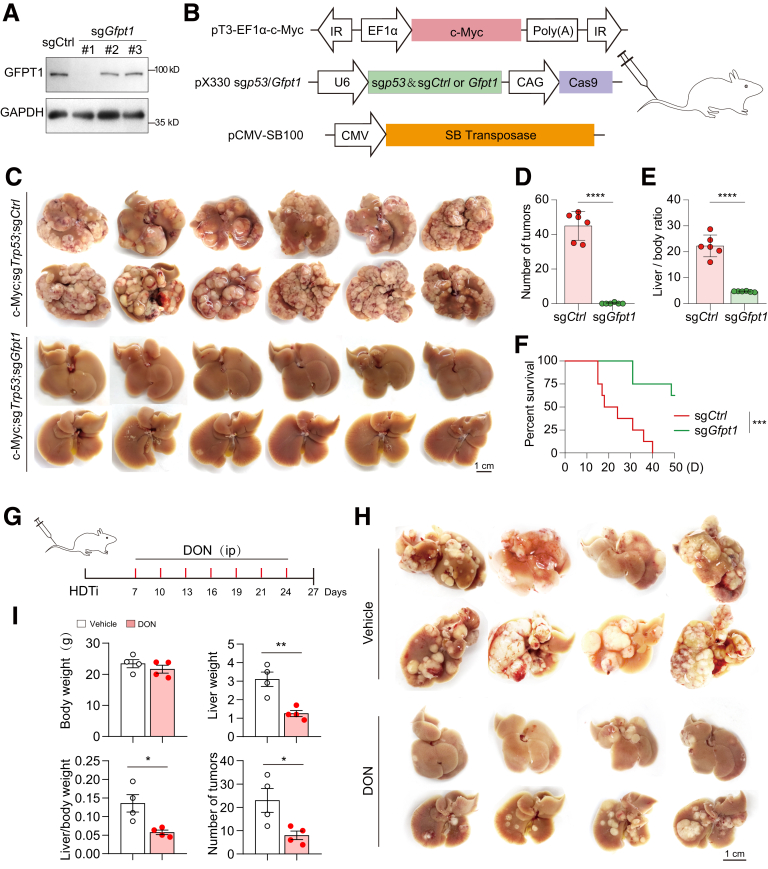
RNA sequencing data revealed that the hexosamine biosynthetic pathway was significantly activated in c-Myc-driven HCC. The rate-limiting enzyme GFPT1 (rather than GFPT2) was up-regulated in the first step of this pathway. Knocking out GFPT1 reduces tumor growth and prolongs murine survival. Human specimens showed that GFPT1 was overexpressed in HCC tissues and was associated with advanced Edmondson-Steiner grades and short patient survival. Further luciferase reporter assays confirmed that c-Myc binds directly to the promoter region of GFPT1 and activates its transcription. Subsequent examination of the downstream pathways of the hexosamine biosynthetic pathway showed that the sialic acid synthesis (but not O-GlcNac glycosylation) pathway was enhanced, which was mediated by a key enzyme, N-acetylneuraminic acid synthase. Knockout of N-acetylneuraminic acid synthase also inhibits tumor growth and extends murine survival in c-Myc-driven HCC models.
These findings indicate that the activation of the hexosamine biosynthetic pathway/sialic acid pathway is an important mechanism underlying the development of c-Myc-driven HCC. Inhibitors of GFPT1, along with anti- N-acetylneuraminic acid synthase may offer a promising therapeutic strategy.
肝细胞癌(HCC)常涉及代谢重编程,这促进了肿瘤发生和转移。然而,其潜在的分子机制仍未得到充分探索。在本研究中,我们旨在研究c-Myc驱动的HCC发展过程中的代谢异常及其潜在的治疗意义。
在通过尾静脉注射c-Myc和sgTrp53/Cas9质粒建立的小鼠HCC模型中,对HCC组织和相邻组织进行RNA测序和代谢组学分析。通过敲除关键催化酶基因来评估肿瘤形成和小鼠存活情况。使用定量聚合酶链反应、免疫组织化学和蛋白质印迹分析基因表达。染色质免疫沉淀后进行定量聚合酶链反应和荧光素酶测定以验证c-Myc调控。
RNA测序数据显示,己糖胺生物合成途径在c-Myc驱动的HCC中显著激活。该途径第一步中的限速酶GFPT1(而非GFPT2)上调。敲除GFPT1可减少肿瘤生长并延长小鼠存活时间。人体标本显示,GFPT1在HCC组织中过表达,且与Edmondson-Steiner分级较高和患者生存期较短相关。进一步的荧光素酶报告基因测定证实,c-Myc直接结合到GFPT1的启动子区域并激活其转录。随后对己糖胺生物合成途径的下游途径进行检查发现,唾液酸合成(而非O-GlcNac糖基化)途径增强,这是由关键酶N-乙酰神经氨酸合酶介导的。敲除N-乙酰神经氨酸合酶也可抑制c-Myc驱动的HCC模型中的肿瘤生长并延长小鼠存活时间。
这些发现表明,己糖胺生物合成途径/唾液酸途径的激活是c-Myc驱动的HCC发展的重要机制。GFPT1抑制剂以及抗N-乙酰神经氨酸合酶可能提供一种有前景的治疗策略。