Grunert Malte, Großmann Max, Hänseroth Jonas, Flötotto Aaron, Oumard Jules, Wolf Johannes Laurenz, Runge Erich, Dreßler Christian
Theoretical Physics I, Institute of Physics, Technische Universität Ilmenau, 98693 Ilmenau, Germany.
Center of Micro- and Nanotechnologies, Technische Universität Ilmenau, 98693 Ilmenau, Germany.
J Phys Chem C Nanomater Interfaces. 2025 May 14;129(21):9662-9669. doi: 10.1021/acs.jpcc.5c02064. eCollection 2025 May 29.
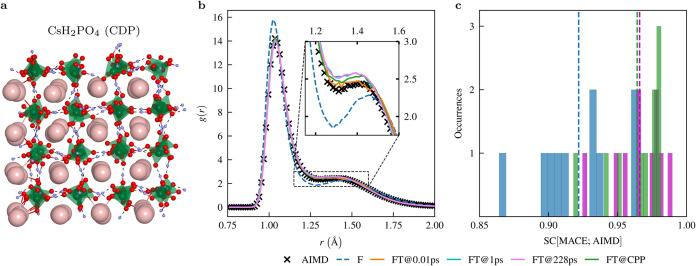
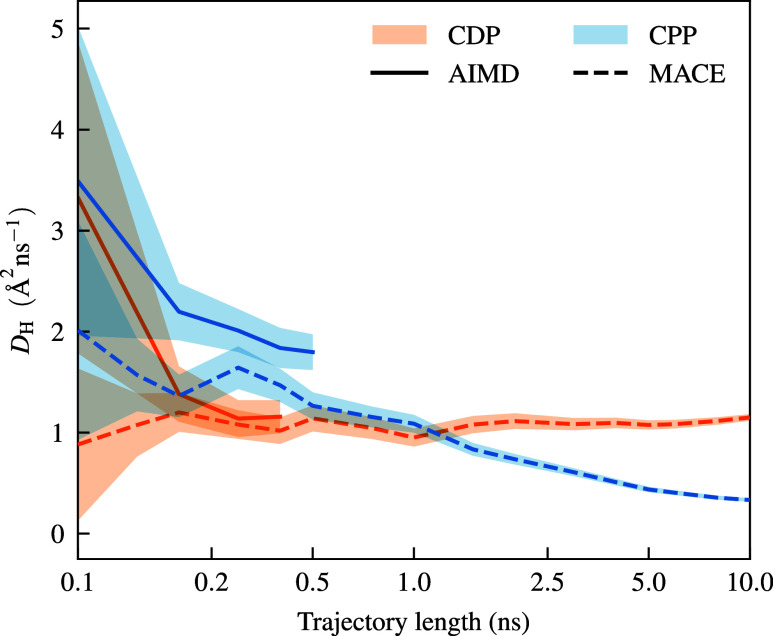
The solid acids CsHPO and Cs(HPO)-(HPO) pose significant challenges for the simulation of proton transport phenomena. In this work, we use the recently developed machine-learned force field (MLFF) MACE to model the proton dynamics on nanosecond time scales for these systems and compare its performance with long-term ab initio molecular dynamics (AIMD) simulations. The MACE-MP-0 foundation model shows remarkable performance for all observables derived from molecular dynamics (MD) simulations, but minor quantitative discrepancies remain compared to the AIMD reference data. However, we show that minimal fine-tuningfitting to as little as 1 ps of AIMD dataleads to full quantitative agreement between the radial distribution functions of MACE force field and AIMD simulations. In addition, we show that traditional long-term AIMD simulations fail to capture the correct qualitative trends in diffusion coefficients and activation energies for these solid acids due to the limited accessible time scale. In contrast, accurate and convergent diffusion coefficients can be reliably obtained through multinanosecond long MD simulations using machine-learned force fields. The obtained qualitative and quantitative behavior of the converged diffusion coefficients and activation energies now matches the experimental trends for both solid acids, in contrast to previous AIMD simulations that yielded a qualitatively wrong picture.
固体酸CsHPO₄和Cs(H₂PO₄)₂(HPO₄)对质子传输现象的模拟提出了重大挑战。在这项工作中,我们使用最近开发的机器学习力场(MLFF)MACE对这些系统在纳秒时间尺度上的质子动力学进行建模,并将其性能与长期的从头算分子动力学(AIMD)模拟进行比较。MACE-MP-0基础模型在所有从分子动力学(MD)模拟得出的可观测量上都表现出卓越的性能,但与AIMD参考数据相比仍存在微小的定量差异。然而,我们表明,只需进行最小程度的微调——仅拟合1皮秒的AIMD数据——就能使MACE力场的径向分布函数与AIMD模拟完全定量一致。此外,我们表明,由于可及的时间尺度有限,传统的长期AIMD模拟无法捕捉这些固体酸在扩散系数和活化能方面正确的定性趋势。相比之下,通过使用机器学习力场进行多纳秒长时间的MD模拟,可以可靠地获得准确且收敛的扩散系数。现在,收敛的扩散系数和活化能所获得的定性和定量行为与两种固体酸的实验趋势相匹配,这与之前得出定性错误结果的AIMD模拟形成对比。