John A. Paulson School of Engineering and Applied Sciences, Harvard University, Cambridge, MA, 02138, USA.
École Polytechnique Fédérale de Lausanne, 1015, Lausanne, Switzerland.
Nat Commun. 2022 May 4;13(1):2453. doi: 10.1038/s41467-022-29939-5.
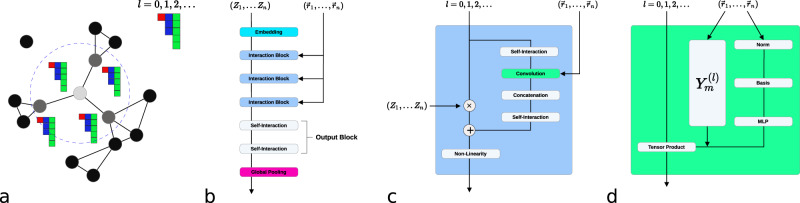
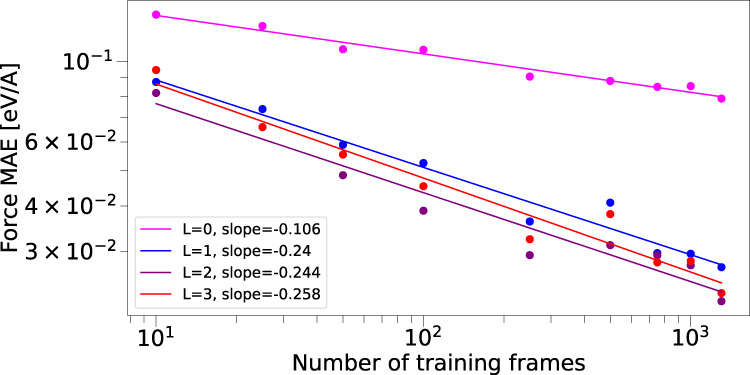
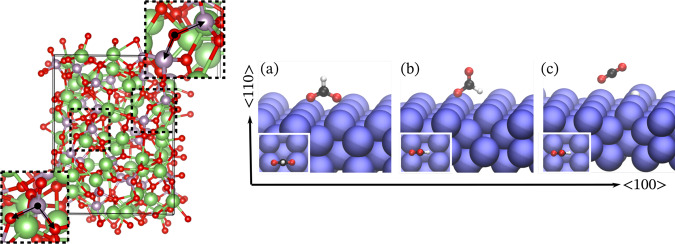
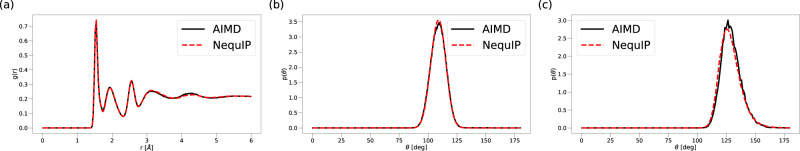
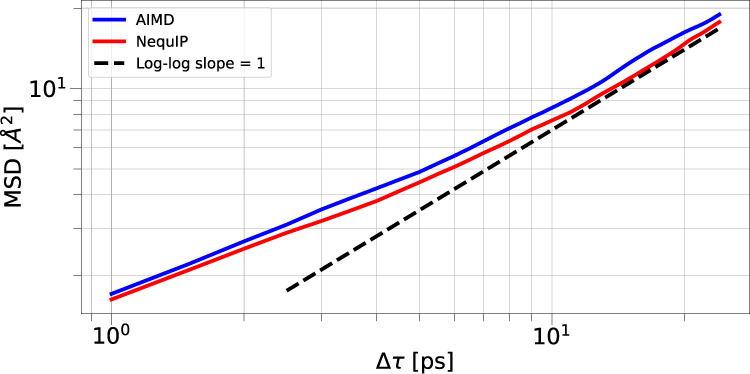
This work presents Neural Equivariant Interatomic Potentials (NequIP), an E(3)-equivariant neural network approach for learning interatomic potentials from ab-initio calculations for molecular dynamics simulations. While most contemporary symmetry-aware models use invariant convolutions and only act on scalars, NequIP employs E(3)-equivariant convolutions for interactions of geometric tensors, resulting in a more information-rich and faithful representation of atomic environments. The method achieves state-of-the-art accuracy on a challenging and diverse set of molecules and materials while exhibiting remarkable data efficiency. NequIP outperforms existing models with up to three orders of magnitude fewer training data, challenging the widely held belief that deep neural networks require massive training sets. The high data efficiency of the method allows for the construction of accurate potentials using high-order quantum chemical level of theory as reference and enables high-fidelity molecular dynamics simulations over long time scales.
本文提出了神经等变相互作用势(NequIP),这是一种从分子动力学模拟的从头算中学习相互作用势的 E(3)等变神经网络方法。虽然大多数现代的对称感知模型使用不变卷积,并且只作用于标量,但 NequIP 采用 E(3)等变卷积来处理几何张量的相互作用,从而更有效地表示原子环境。该方法在一组具有挑战性和多样性的分子和材料上实现了最先进的准确性,同时表现出显著的数据效率。NequIP 用多达三个数量级更少的训练数据超越了现有模型,挑战了深度学习网络需要大规模训练集的普遍观点。该方法的数据效率很高,可以使用高精度量子化学理论作为参考构建准确的势能,并能够在长时间尺度上进行高保真的分子动力学模拟。