Machine Learning Group, Technische Universität Berlin, 10587 Berlin, Germany.
Physics and Materials Science Research Unit, University of Luxembourg, L-1511 Luxembourg, Luxembourg.
Sci Adv. 2017 May 5;3(5):e1603015. doi: 10.1126/sciadv.1603015. eCollection 2017 May.
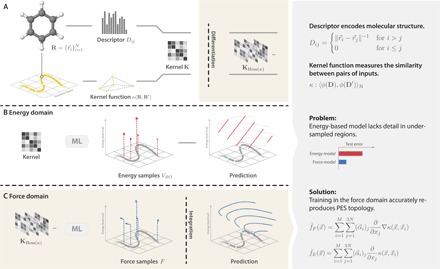
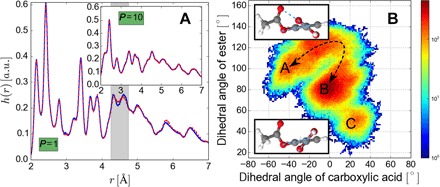
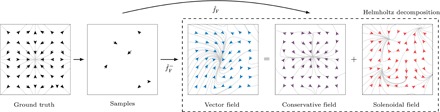
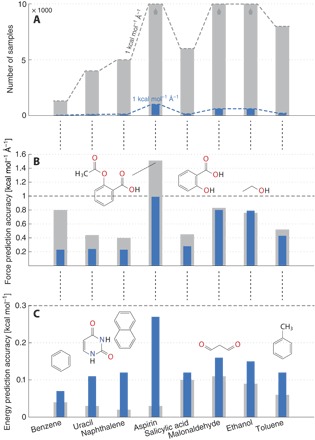
Using conservation of energy-a fundamental property of closed classical and quantum mechanical systems-we develop an efficient gradient-domain machine learning (GDML) approach to construct accurate molecular force fields using a restricted number of samples from ab initio molecular dynamics (AIMD) trajectories. The GDML implementation is able to reproduce global potential energy surfaces of intermediate-sized molecules with an accuracy of 0.3 kcal mol for energies and 1 kcal mol Å̊ for atomic forces using only 1000 conformational geometries for training. We demonstrate this accuracy for AIMD trajectories of molecules, including benzene, toluene, naphthalene, ethanol, uracil, and aspirin. The challenge of constructing conservative force fields is accomplished in our work by learning in a Hilbert space of vector-valued functions that obey the law of energy conservation. The GDML approach enables quantitative molecular dynamics simulations for molecules at a fraction of cost of explicit AIMD calculations, thereby allowing the construction of efficient force fields with the accuracy and transferability of high-level ab initio methods.
利用能量守恒——封闭经典和量子力学系统的基本属性——我们开发了一种高效的梯度域机器学习 (GDML) 方法,通过从从头分子动力学 (AIMD) 轨迹中获取有限数量的样本,构建精确的分子力场。GDML 实现能够使用仅 1000 个构象几何形状进行训练,以 0.3 kcal/mol 的精度重现中等大小分子的全局势能表面,以 1 kcal/mol Å 的精度重现原子力。我们在包括苯、甲苯、萘、乙醇、尿嘧啶和阿司匹林在内的分子的 AIMD 轨迹中证明了这一准确性。通过在满足能量守恒定律的向量值函数的 Hilbert 空间中学习,我们的工作完成了构建保守力场的挑战。GDML 方法能够以显式 AIMD 计算成本的一小部分进行分子的定量动力学模拟,从而能够以高精度和高级从头算方法的可转移性构建高效的力场。