Bovo Samuele, Ribani Anisa, Schiavo Giuseppina, Taurisano Valeria, Bolner Matteo, Bertolini Francesca, Fontanesi Luca
Animal and Food Genomics Group, Division of Animal Sciences, Department of Agricultural and Food Sciences, University of Bologna, 40127 Bologna, Italy.
Vet Sci. 2025 May 24;12(6):513. doi: 10.3390/vetsci12060513.
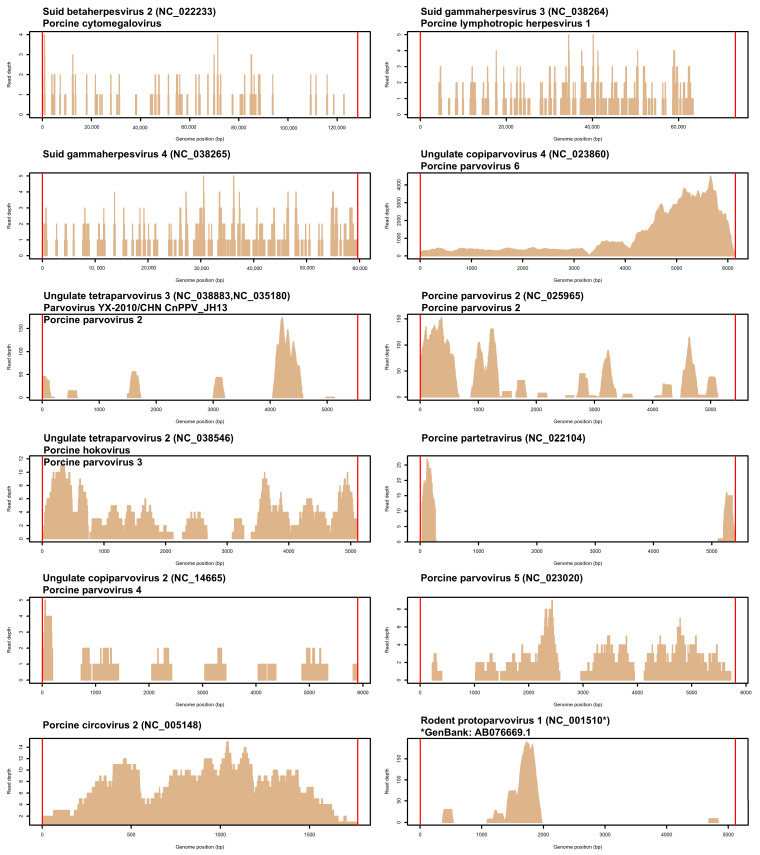
Pigs are affected by a variety of pathogenic agents that need to be identified correctly and diagnosed even when co-infections may complicate the application of specific and targeted assays. Next-generation sequencing can provide new perspective to monitor viruses infecting or co-infecting diseased pigs. In this study, we tested, for the first time for diagnostic purposes in a livestock species, a new method based on whole-genome sequencing of all the DNAs extracted from the blood of nine pigs sampled from a farm where there was a suspected outbreak of Post-weaning Multisystemic Wasting Syndrome. We then used unmapped reads on the porcine reference genome to mine for viral sequences using a specifically designed bioinformatic pipeline. Within this fraction of reads, viral sequences ranged from 0.002% to 4.4% of the total unmapped reads and were derived from twelve different viruses known to infect pigs, where three were herpesviruses, eight were parvoviruses, and one was a circovirus. All pig sequencing datasets were positive for one or more viruses, with various potential viral loads. Suid betaherpesvirus 2, also known as Porcine cytomegalovirus (PCMV), was the most frequently identified virus as five out of the nine pig sequencing datasets contained viral sequences from this virus. The results may suggest a heterogeneous viral profile of the diseased pigs that may be derived from potential secondary infections or co-infections. This pilot application demonstrated that a whole-genome sequencing approach can complement other routine diagnostic assays in veterinary virology. Other studies and improvements are needed to validate the results and apply this approach in routine monitoring applications.
猪会受到多种致病因子的影响,即使合并感染可能会使特异性和靶向性检测方法的应用变得复杂,也需要正确识别和诊断这些致病因子。新一代测序技术可以为监测感染患病猪或与患病猪共同感染的病毒提供新的视角。在本研究中,我们首次出于诊断目的,对从一个疑似发生断奶后多系统消耗综合征疫情的农场采集的9头猪的血液中提取的所有DNA进行全基因组测序,测试了一种新方法。然后,我们使用猪参考基因组上未映射的 reads,通过专门设计的生物信息学流程来挖掘病毒序列。在这部分 reads 中,病毒序列占未映射 reads 总数的0.002%至4.4%,来源于已知感染猪的12种不同病毒,其中3种是疱疹病毒,8种是细小病毒,1种是圆环病毒。所有猪的测序数据集都对一种或多种病毒呈阳性,病毒载量各不相同。猪β疱疹病毒2,也称为猪巨细胞病毒(PCMV),是最常被鉴定出的病毒,9个猪测序数据集中有5个包含该病毒的病毒序列。结果可能表明患病猪的病毒谱具有异质性,这可能源于潜在的继发感染或合并感染。这项初步应用表明,全基因组测序方法可以补充兽医病毒学中的其他常规诊断检测方法。需要进行其他研究和改进,以验证结果并将这种方法应用于常规监测应用中。