Gerezgiher Kidane Goitom, Tafere Dessie Ashagrie, Teklehaimanot Tesfay Gebremikael, Tadesse Ashenafi Belihu, Dinkirie Ayalew Manahilie, Zeweldi Hagos Woldeghebriel
Department of Physics, CNCS, Mekelle University P.O. Box 231 Mekelle Ethiopia
Department of Chemistry College of Natural and Computational Science, Mekdela Amba University P.O. Box 32 Tuluawulia Ethiopia.
RSC Adv. 2025 Jul 1;15(27):21541-21554. doi: 10.1039/d5ra03371c. eCollection 2025 Jun 23.
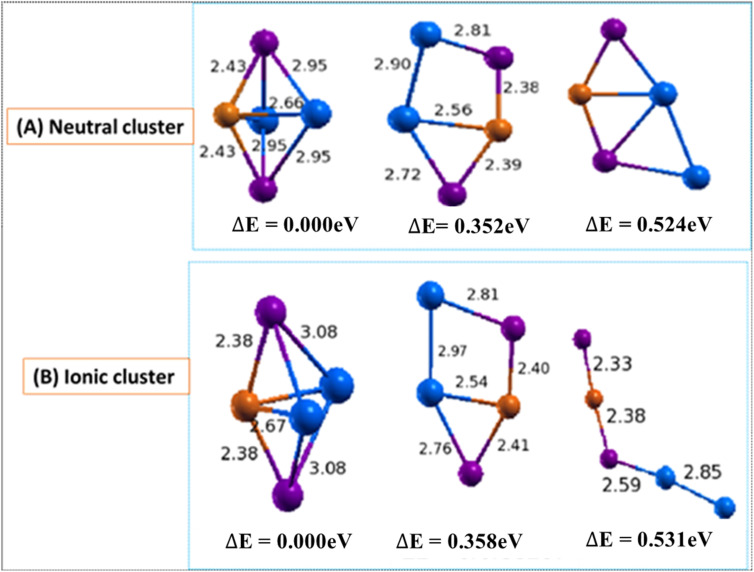
In this work, the electronic and structural properties of Cu In Te neutral and anion clusters are studied. The simulations are carried out using the QUANTUM ESPRESSO/PWSCF package, based on the density functional theory (DFT) principle, which employs a pseudo-potential with a plane wave basis set. Geometry optimization starting from several initial candidate structures was performed for each cluster size to determine the number of possible minimum-energy isomers for each size. The results show that the lowest-energy structures are cubic, ranging from cluster = 2 to 5, and resemble the chalcopyrite structure. The geometry of neutral and anionic cases exhibits a structural change, including distortion and a transition from two-dimensional to one-dimensional. By considering energetics, HOMO-LUMO gap, binding energy, ionization potential and electron affinity, the relative stability of Cu In Te/(Cu In Te) was measured. From the most stable energy structures, CuInTe/(CuInTe) were found to have enhanced chemical stability relative to their neighbours. They are a magic-number species. The binding energy and HOMO-LUMO gap of CuInTe/(CuInTe) clusters show the most significant value, which indicates high chemical stability. The adiabatic ionization potential of the cluster decreases monotonically, showing favor for metallic character as cluster size increases. Both clusters' vertical/adiabatic detachment energies also show a slight odd-even oscillation with an increasing tendency as a function of cluster size. This indicates that the successive increase in metallic atoms results in a decrease in nonmetallic favor. We also analyse the partial charge density of the optimized geometries for both anion and neutral clusters. The numerical value indicates that these clusters, including photovoltaic solar cells and other devices, make a significant contribution to semiconductor design.
在这项工作中,研究了CuInTe中性和阴离子团簇的电子及结构性质。基于密度泛函理论(DFT)原理,使用QUANTUM ESPRESSO/PWSCF软件包进行模拟,该软件包采用赝势平面波基组。针对每个团簇尺寸,从几种初始候选结构开始进行几何优化,以确定每种尺寸下可能的最低能量异构体数量。结果表明,最低能量结构为立方结构,团簇尺寸从2到5,类似于黄铜矿结构。中性和阴离子情况的几何结构呈现出结构变化,包括畸变以及从二维到一维的转变。通过考虑能量学、HOMO-LUMO能隙、结合能、电离势和电子亲和势,测量了CuInTe/(CuInTe)的相对稳定性。从最稳定的能量结构来看,发现CuInTe/(CuInTe)相对于其相邻结构具有增强的化学稳定性。它们是幻数物种。CuInTe/(CuInTe)团簇的结合能和HOMO-LUMO能隙显示出最显著的值,这表明其具有高化学稳定性。团簇的绝热电离势单调下降,表明随着团簇尺寸增加,其金属特性增强。两个团簇的垂直/绝热脱离能也随着团簇尺寸增加呈现出轻微的奇偶振荡且有增加趋势。这表明金属原子的连续增加导致非金属特性降低。我们还分析了阴离子和中性团簇优化几何结构的部分电荷密度。数值表明,这些团簇,包括光伏太阳能电池和其他器件,对半导体设计有重大贡献。