Priyadharshini M, Raju B Deevena, Banu A Faritha, Kumar P Jagdish, Murugesh V, Rybin Oleg
Department of Computer Science & Engineering, Faculty of Science and Technology (IcfaiTech), ICFAI Foundation for Higher Education, Hyderabad, 501203, India.
Department of Artificial Intelligence & Data Science, Faculty of Science and Technology (IcfaiTech), ICFAI Foundation for Higher Education, Hyderabad, 501203, India.
Sci Rep. 2025 Jul 22;15(1):26553. doi: 10.1038/s41598-025-06544-2.
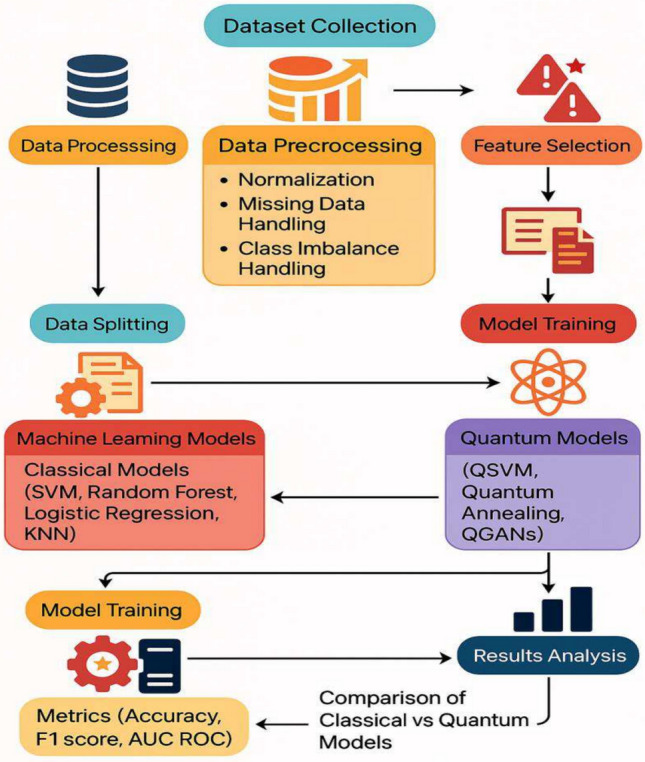
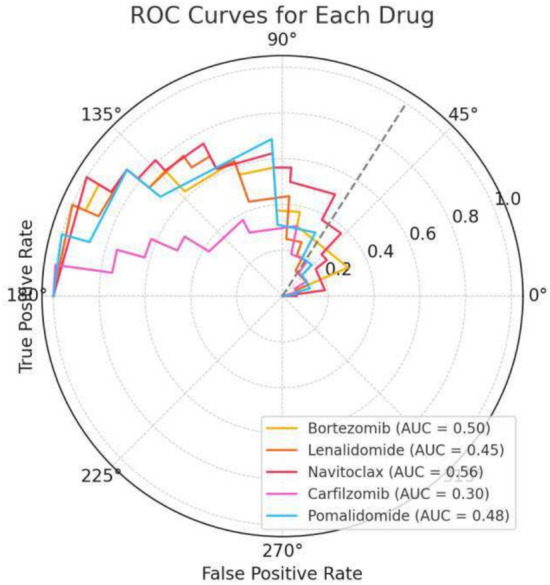
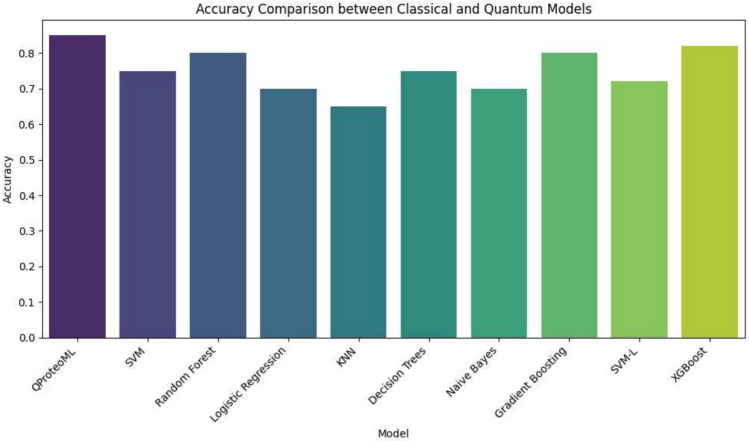
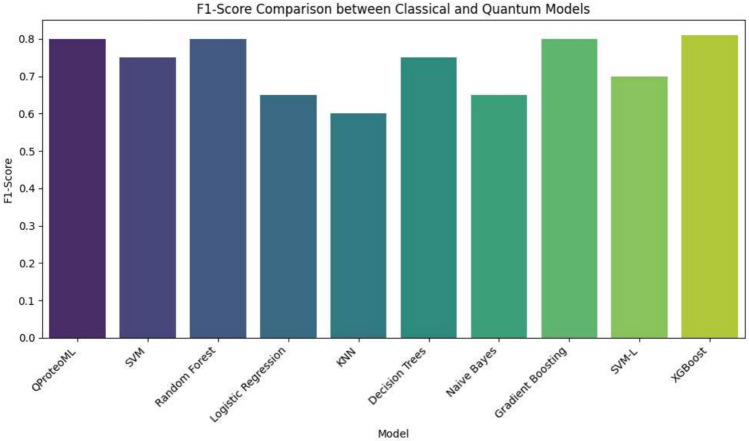
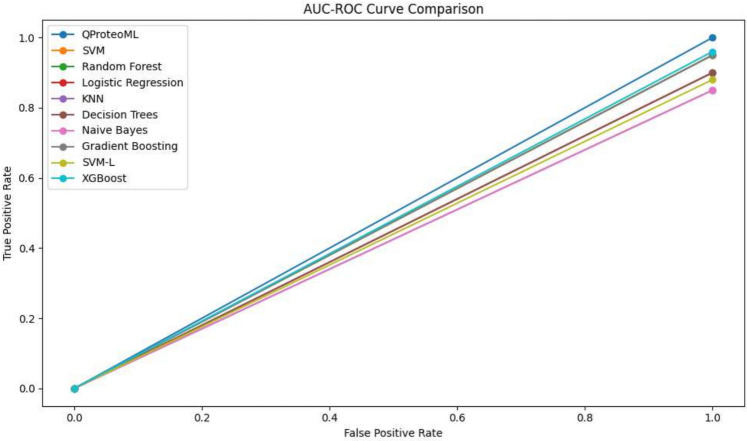
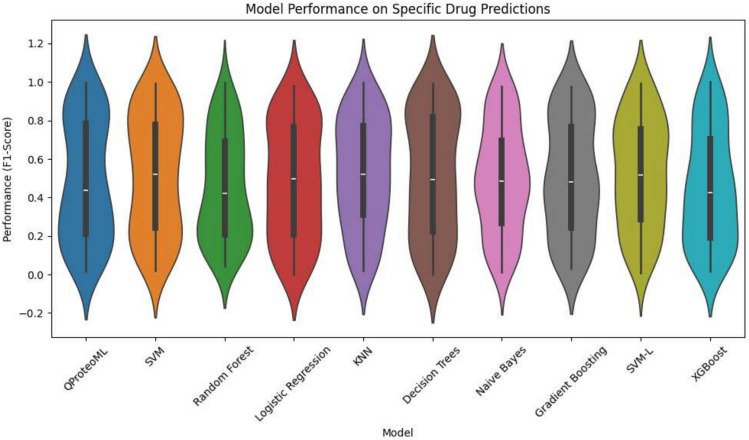
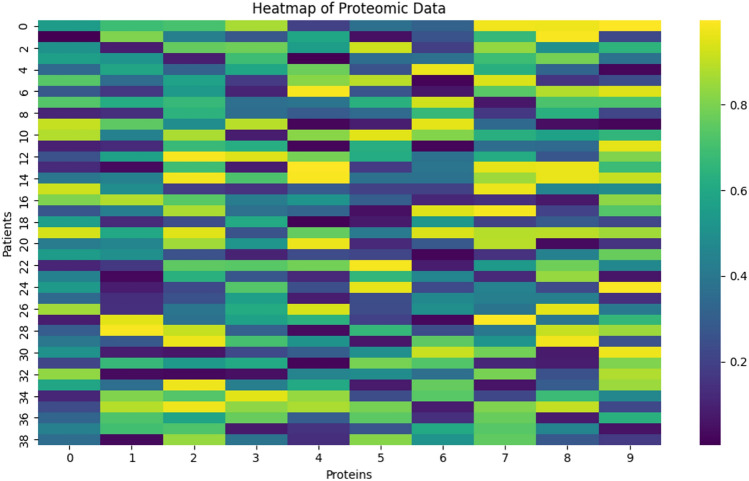
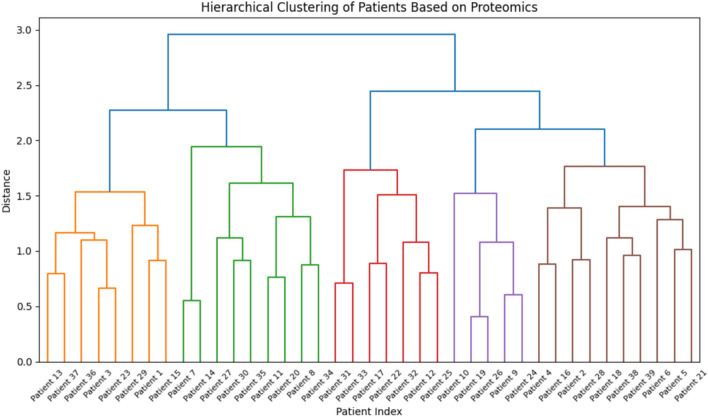
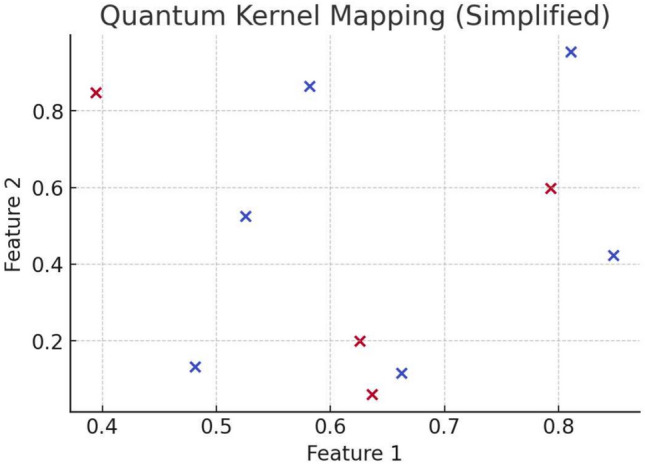
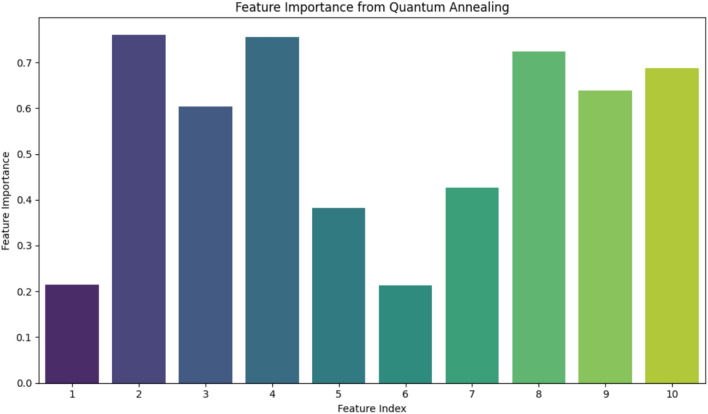
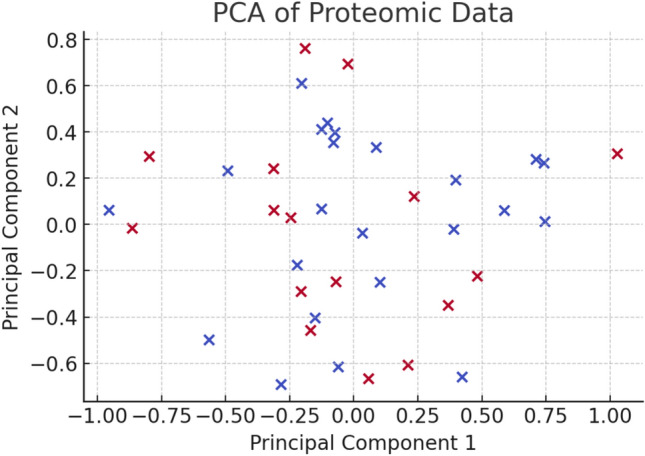
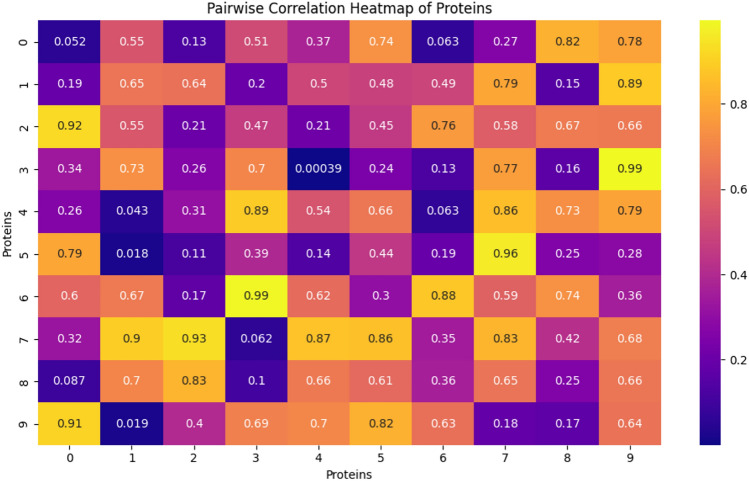
In this paper, we introduce QProteoML, a new quantum machine learning (QML) framework for predicting drug sensitivity in Multiple Myeloma (MM) using high-dimensional proteomic data. MM, an extremely heterogeneous condition, displays often mixed responses to treatment, with a large number of patients showing drug resistance to proteasome inhibitors and immune modulatory agents. However, the methods previously used for genomic and proteomic data analysis techniques are plagued by issues of high dimensionality, imbalanced class distribution and feature redundancy, which work against the accurate predictability and generalizability of such methods. These are compounded by the so-called "curse of dimensionality", with dimensions far outnumbering samples, hence classical model overfitting. In this work, we present QProteoML as an integration of quantum techniques purposefully developed to deal with high-dimensional, imbalanced and redundant data. The framework integrates a combination of Quantum Support Vector Machine (QSVM), Quantum Principal Component Analysis (qPCA), Quantum Annealing (QA) for feature selection and Quantum Generative Adversarial Networks (QGANs) for data augmentation. These quantum algorithms exploit certain quantum phenomena (superposition and entanglement) to perform modelling of nonlinear relationships, dimensionality reduction, and class-imbalance issues. QSVM employs quantum kernels to map data into a higher-dimensional Hilbert space, so that the model can detect complex patterns in MM drug resistance. qPCA reduces dimensionality without loss of important variance, and thus improves computation efficiency. In addition, Quantum Annealing successfully extracts the most informative biomarkers with low redundancy. QProteoML was experimentally tested by comparing accuracy, F1 score and AUC ROC between classical machine learning models such as Support Vector Machine (SVM), Random Forest (RF), Logistic Regression (LR), and K-Nearest Neighbors (KNN). Our results demonstrate that QProteoML performs better than classical models, particularly in identifying the drug resistant minority class of patients. Additionally, the model is interpretable and stresses important biomarkers of drug sensitivity in MM. This research opens the possibility of quantum machine learning in personalised medicine for Multiple Myeloma. It demonstrates that quantum algorithms can perform complex biological data suggesting more reliable and accurate drug sensitivity predictions. Future research will be directed toward clinical validation of the given system with larger and more diverse cohorts of MM patients; the integration of quantum hardware for practical applications.
在本文中,我们介绍了QProteoML,这是一种新的量子机器学习(QML)框架,用于使用高维蛋白质组数据预测多发性骨髓瘤(MM)中的药物敏感性。MM是一种极其异质性的疾病,对治疗的反应往往参差不齐,大量患者对蛋白酶体抑制剂和免疫调节剂表现出耐药性。然而,先前用于基因组和蛋白质组数据分析技术的方法受到高维度、类分布不平衡和特征冗余等问题的困扰,这些问题不利于此类方法的准确预测性和通用性。这些问题因所谓的“维度诅咒”而更加复杂,维度数量远远超过样本数量,从而导致经典模型过度拟合。在这项工作中,我们提出QProteoML,它是为处理高维、不平衡和冗余数据而专门开发的量子技术的集成。该框架集成了量子支持向量机(QSVM)、量子主成分分析(qPCA)、用于特征选择的量子退火(QA)和用于数据增强的量子生成对抗网络(QGAN)。这些量子算法利用某些量子现象(叠加和纠缠)来进行非线性关系建模、降维和类不平衡问题处理。QSVM使用量子核将数据映射到更高维的希尔伯特空间,以便模型能够检测MM耐药性中的复杂模式。qPCA在不损失重要方差的情况下降低维度,从而提高计算效率。此外,量子退火成功提取了冗余度低的最具信息性的生物标志物。通过比较支持向量机(SVM)、随机森林(RF)、逻辑回归(LR)和K近邻(KNN)等经典机器学习模型的准确率、F1分数和AUC ROC,对QProteoML进行了实验测试。我们的结果表明,QProteoML的性能优于经典模型,特别是在识别耐药性少数患者群体方面。此外,该模型具有可解释性,并强调了MM中药物敏感性的重要生物标志物。这项研究开启了量子机器学习在多发性骨髓瘤个性化医疗中的可能性。它表明量子算法可以处理复杂的生物数据,从而做出更可靠、准确的药物敏感性预测。未来的研究将朝着使用更大、更多样化的MM患者队列对给定系统进行临床验证;以及将量子硬件集成到实际应用中。