Delabrière Alexis, Gianfrotta Coline, Dechaumet Sylvain, Damont Annelaure, Hautbergue Thaïs, Roger Pierrick, Jamin Emilien L, Puel Olivier, Junot Christophe, Fenaille François, Thévenot Etienne A
Université Paris-Saclay, CEA, List, Palaiseau, France.
Département Médicaments et Technologies pour la Santé, Université Paris-Saclay, CEA, INRAE, Gif-sur-Yvette, France.
J Cheminform. 2025 Jul 24;17(1):111. doi: 10.1186/s13321-025-01051-y.
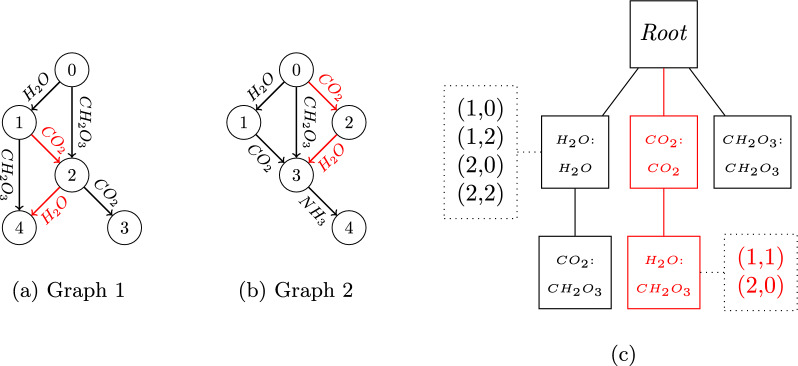
Identification is a major challenge in metabolomics due to the large structural diversity of metabolites. Tandem mass spectrometry is a reference technology for studying the fragmentation of molecules and characterizing their structure. Recent instruments can fragment large amounts of compounds in a single acquisition. The search for similarities within a collection of MS/MS spectra is a powerful approach to facilitate the identification of new metabolites. We propose an innovative de novo strategy for searching for exact fragmentation patterns within collections of MS/MS spectra. This approach is based on (i) a new representation of spectra as graphs of m/z differences, and (ii) an efficient frequent-subgraph mining algorithm. We demonstrate both on a spectral database from standards and on acquisitions in biological matrices that these new fragmentation patterns capture similarities that are not extracted by existing methods, and facilitate the structural interpretation of molecular network components and the elucidation of unknown spectra. The mineMS2 software is publicly available as an R package ( https://github.com/odisce/mineMS2 ). SCIENTIFIC CONTRIBUTION: We present an innovative strategy for structural elucidation, which extracts exact fragmentation patterns of m/z differences within collections of MS/MS spectra. The algorithms are implemented in a software library enabling efficient mining of MS/MS data and coupling to molecular networks. We show on real datasets the specific value of the patterns as fragmentation graphs for structural interpretation and de novo identification, and their complementarity to existing approaches.
由于代谢物的结构多样性极大,鉴定成为代谢组学中的一项重大挑战。串联质谱是研究分子裂解并表征其结构的参考技术。最新的仪器能够在一次采集过程中裂解大量化合物。在MS/MS谱图集合中寻找相似性是促进新代谢物鉴定的一种有效方法。我们提出了一种创新的从头搜索策略,用于在MS/MS谱图集合中寻找精确的裂解模式。该方法基于:(i)将谱图表示为质荷比差异图的新方法,以及(ii)一种高效的频繁子图挖掘算法。我们在标准物质的谱图数据库以及生物基质的采集数据上均证明,这些新的裂解模式能够捕捉现有方法无法提取的相似性,有助于分子网络成分的结构解析以及未知谱图的阐释。mineMS2软件作为R包(https://github.com/odisce/mineMS2)可公开获取。科学贡献:我们提出了一种创新的结构解析策略,该策略可在MS/MS谱图集合中提取质荷比差异的精确裂解模式。这些算法在一个软件库中实现,能够高效挖掘MS/MS数据并与分子网络相结合。我们在实际数据集上展示了这些模式作为裂解图在结构解析和从头鉴定方面的特殊价值,以及它们与现有方法的互补性。