Vlad Raluca Maria, Dijmărescu Irina, Dobritoiu Ruxandra, Moga Andreea, Balanescu Laura, Neagu Oana, Pacurar Daniela
Department of Paediatrics, "Carol Davila" University of Medicine and Pharmacy, 020021 Bucharest, Romania.
"Grigore Alexandrescu" Emergency Children's Hospital, 011743 Bucharest, Romania.
Reports (MDPI). 2025 Mar 17;8(1):33. doi: 10.3390/reports8010033.
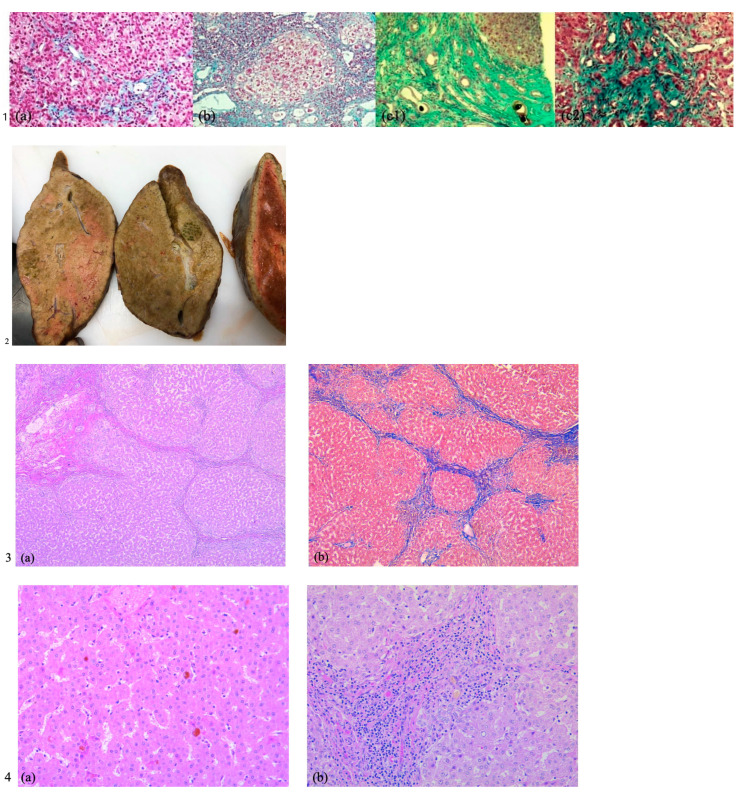
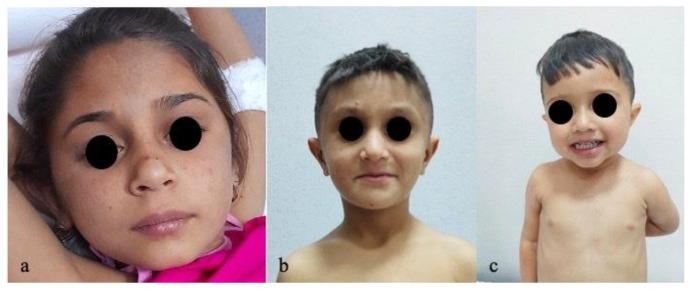
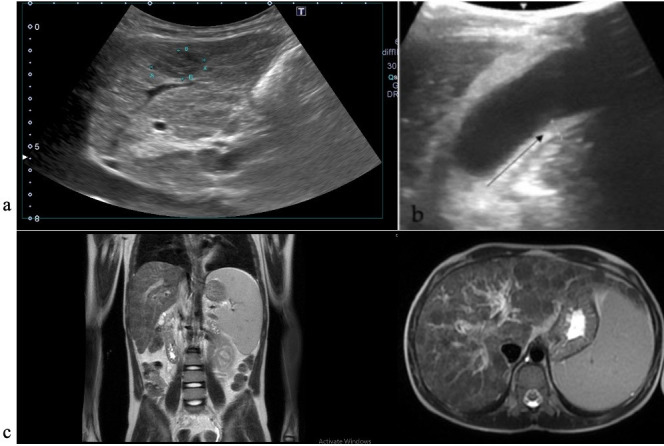
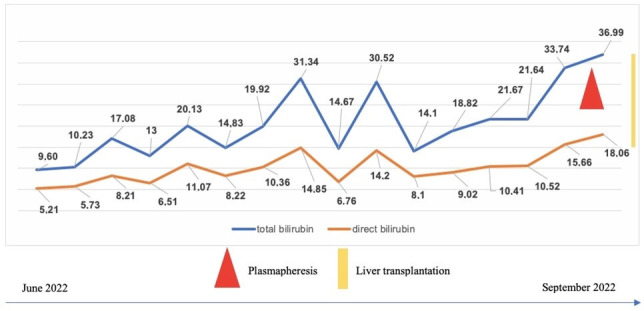
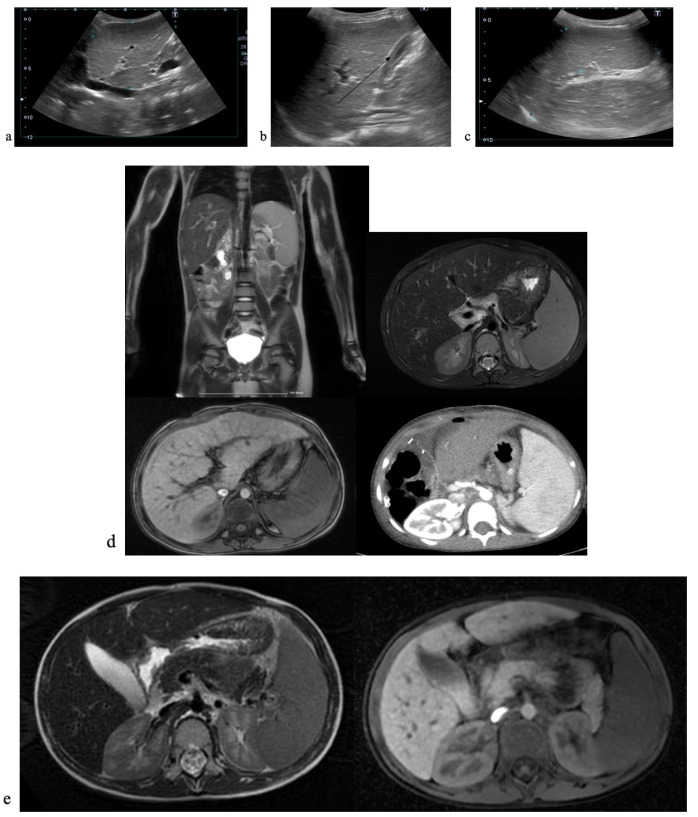
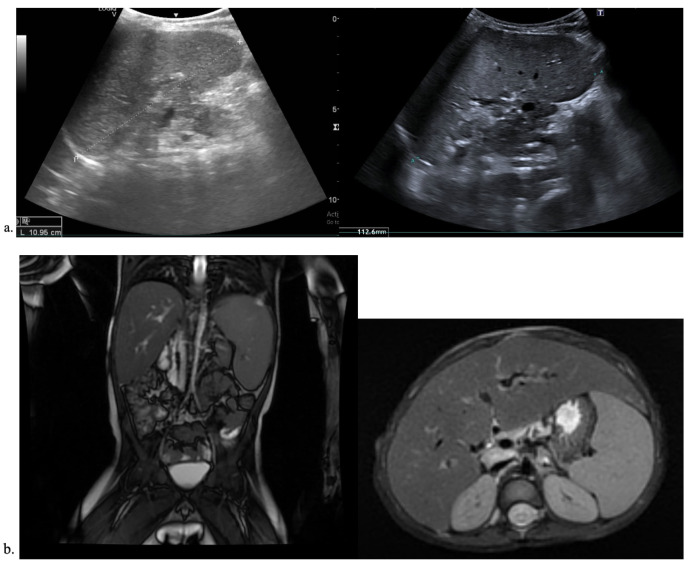
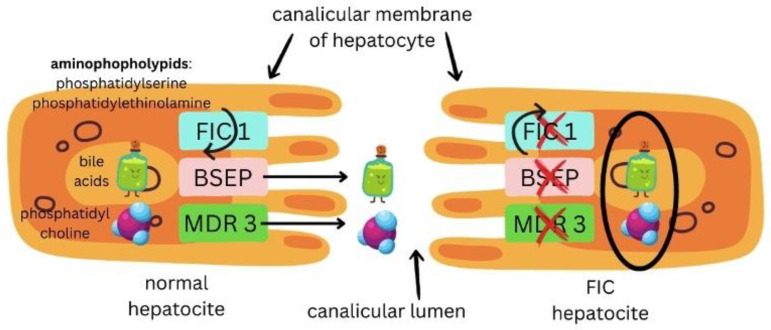
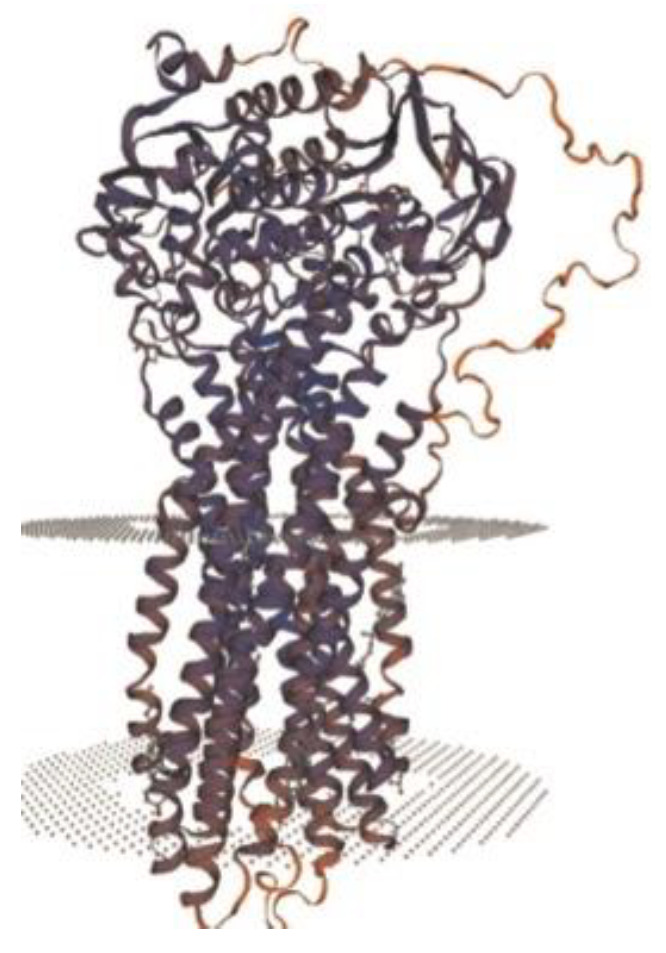
Progressive familial intrahepatic cholestasis (PFIC) refers to a heterogeneous group of autosomal recessive disorders consisting of mutations of hepatocyte transporting-system genes involved in bile formation. The exact prevalence remains unknown but is estimated at 1 in 500.000 for PFIC 3, caused by mutations in the ABCB4 gene. We report three cases of PFIC 3 from the patient's sister, brother, and cousin, diagnosed in our Pediatric Department in 2022-2023. Case 1: A 10-year-old girl was admitted for jaundice and abdominal pain. She was diagnosed with severely advanced hepatic cirrhosis and massive cholestasis. Genetic testing showed ABCB4 homozygous mutation. She rapidly developed fulminant liver failure, and a living donor liver transplant was performed. Case 2: A 6-year-old brother was previously diagnosed with cholestatic hepatitis of unknown cause back in 2018 and presented with similar features (generalized jaundice, severe pruritus with generalized scratching lesions); symptoms had progressively developed from the first year of life. He also exhibited particular facial features (big forehead, twisted ear lobe, straight nose). He received cadaveric liver transplantation. Case 3: Nephew of first two children, a 3-year-5-month-old boy, was admitted for failure to thrive and a one-year history of jaundice, pruritus, and splenomegaly. He was tested positive for homozygous ABCB4 mutation. He is currently under medical treatment with stable liver function. The clinical significance of this particular homozygous variant identified in ABCB4 in our series of cases (c.2534G>T (p.Gly845Val)) was uncertain up to this case report. The present data provide convincing evidence as to the correlation between this mutation and the clinical phenotype of PFIC 3.
进行性家族性肝内胆汁淤积症(PFIC)是一组异质性常染色体隐性疾病,由参与胆汁形成的肝细胞转运系统基因突变引起。确切患病率尚不清楚,但据估计,由ABCB4基因突变导致的PFIC 3患病率为1/500000。我们报告了2022年至2023年在我们儿科诊断出的3例PFIC 3病例,分别来自患者的姐姐、哥哥和表弟。病例1:一名10岁女孩因黄疸和腹痛入院。她被诊断为严重晚期肝硬化和大量胆汁淤积。基因检测显示ABCB4纯合突变。她迅速发展为暴发性肝衰竭,并接受了活体供肝移植。病例2:一名6岁男孩早在2018年就被诊断为病因不明的胆汁淤积性肝炎,表现出类似症状(全身黄疸、严重瘙痒伴全身抓痕性病变);症状自出生第一年起逐渐发展。他还表现出特殊的面部特征(额头大、耳垂扭曲、鼻梁挺直)。他接受了尸体肝移植。病例3:前两个孩子的侄子,一名3岁5个月大的男孩,因发育不良以及有1年黄疸、瘙痒和脾肿大病史入院。他ABCB4纯合突变检测呈阳性。他目前正在接受治疗,肝功能稳定。在我们的系列病例中鉴定出的ABCB4中这种特定的纯合变异体(c.2534G>T(p.Gly845Val))的临床意义在本病例报告之前尚不确定。目前的数据为这种突变与PFIC 3临床表型之间的相关性提供了令人信服的证据。