Shrestha Ram Lal Swagat, M C Shiva, Tamang Ashika, Poudel Manila, Parajuli Nirmal, Shrestha Aakar, Shrestha Timila, Bharati Samjhana, Maharjan Binita, Marasini Bishnu P, Adhikari Subin Jhashanath
Department of Chemistry, Amrit Campus, Tribhuvan University, Lainchaur, Kathmandu, Nepal.
Kathmandu Valley College, Kalanki, Kathmandu, Nepal.
PLoS One. 2025 Jul 30;20(7):e0329168. doi: 10.1371/journal.pone.0329168. eCollection 2025.
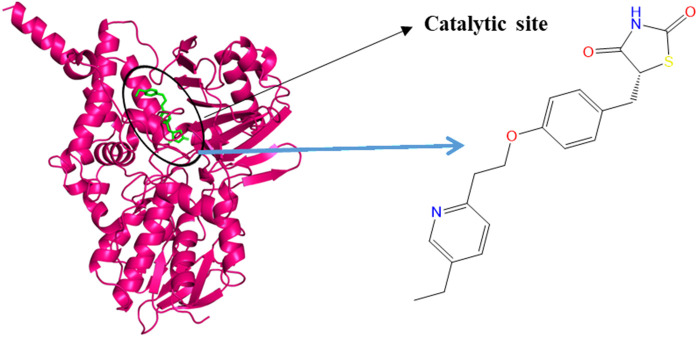
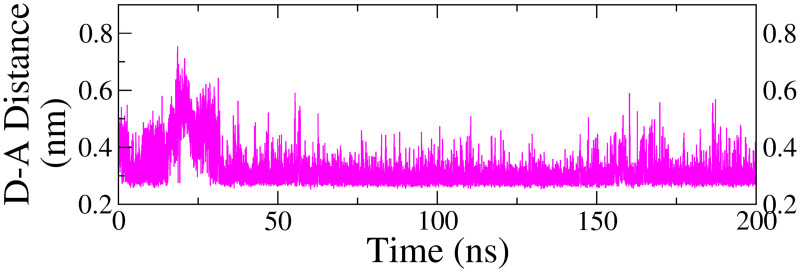
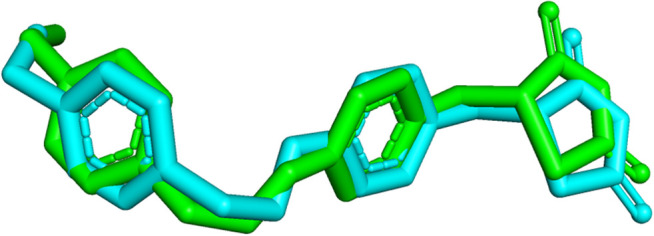
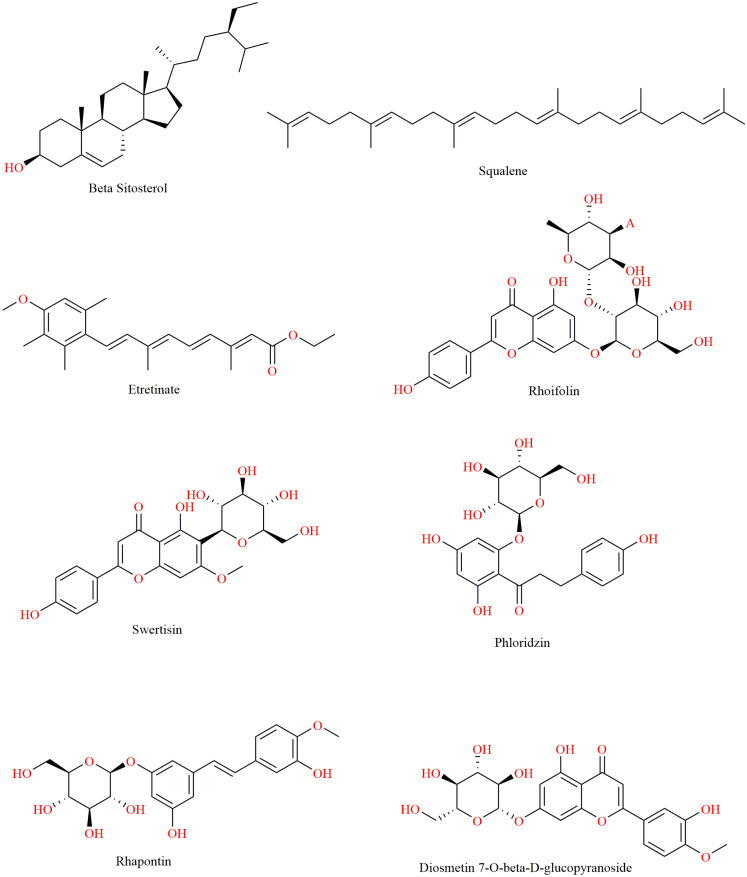
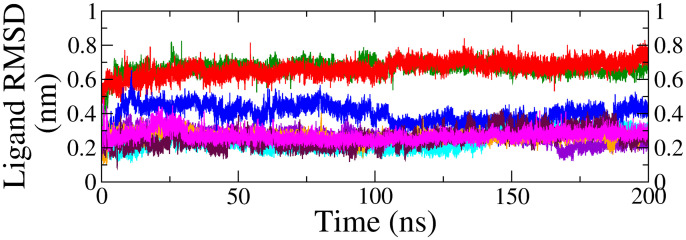
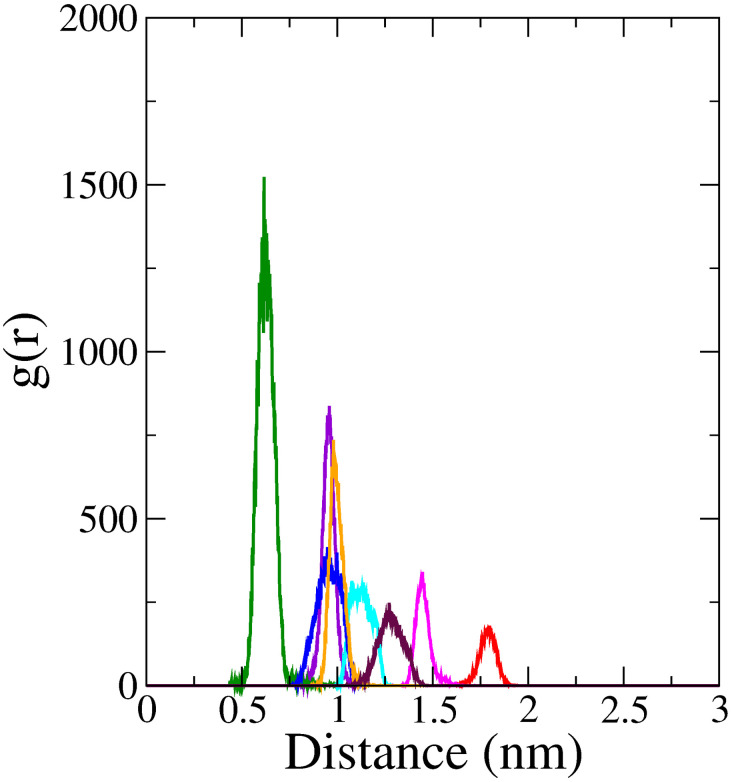
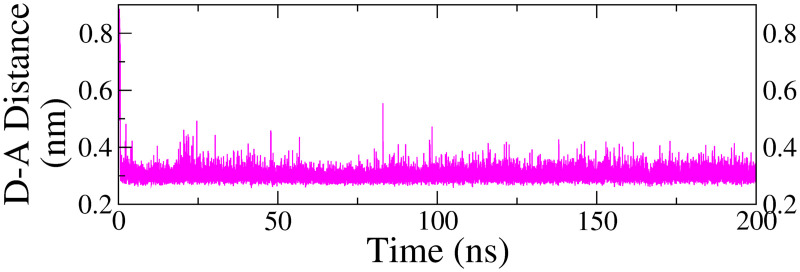
Monoamine oxidase B (MAO-B) serves as a critical target in the management of neurodegenerative diseases (NDDs) such as Alzheimer's and Parkinson's due to its role in regulating oxidative stress and dopamine metabolism. In this context, phytochemicals from Oxalis species, known for their neuroprotective properties, were explored for their potential MAO-B inhibitory activity using computational approach. Plant-derived compounds, offering a better safety profile than synthetic drugs and greater cost-effectiveness, present a promising avenue for developing alternative therapeutic strategies. Molecular docking (MD), molecular dynamics simulations (MDS), and binding free energy calculations were employed to evaluate the inhibitory potential of Oxalis phytochemicals against MAO-B (PDB ID: 4A79). Stable ligand-protein complexes with optimal docking scores were selected, and key parameters from molecular dynamics trajectories, including binding stability and interactions, were analyzed to identify high potential inhibitors of MAO-B for therapeutic development. Results showed beta-sitosterol (-11.92 kcal/mol), squalene (-11.89 kcal/mol), etretinate (-11.46 kcal/mol), rhoifolin (-11.44 kcal/mol), and swertisin (-11.13 kcal/mol) demonstrated superior binding affinities compared to the native ligand (-11.12 kcal/mol). Three additional compounds; phloridzin (-11.10 kcal/mol), rhapontin (-11.02 kcal/mol), and diosmetin 7-O-beta-D-glucopyranoside (-10.96 kcal/mol) exhibited better binding than reference drugs. The predominant interactions between protein and ligand were hydrophobic, with hydrogen bonds and Pi-stacking enhancing the complexes' stability. The evaluation based on geometrical and thermodynamic metrics derived from 200 ns MDS, identified rhoifolin, beta-sitosterol, and swertisin as promising MAO-B inhibitors. Minimal translational and rotational movements of these ligands within the catalytic site of MAO-B under quasi-physiological conditions suggested effective inhibition. Preserved thermodynamic feasibility reinforced these findings. ADMET analysis identified squalene and beta-sitosterol as CNS active candidates with favorable pharmacokinetics, while etretinate, rhoifolin, and swertisin may act as peripheral modulators, requiring optimization for improved CNS delivery. Further experimental validation of efficacy, pharmacokinetics, and safety is recommended to advance the therapeutic potential of these hit candidates.
单胺氧化酶B(MAO-B)由于在调节氧化应激和多巴胺代谢中发挥作用,成为治疗阿尔茨海默病和帕金森病等神经退行性疾病(NDDs)的关键靶点。在此背景下,利用计算方法探索了酢浆草属植物中具有神经保护特性的植物化学物质的潜在MAO-B抑制活性。植物来源的化合物具有比合成药物更好的安全性和更高的成本效益,为开发替代治疗策略提供了一条有前景的途径。采用分子对接(MD)、分子动力学模拟(MDS)和结合自由能计算来评估酢浆草植物化学物质对MAO-B(PDB ID:4A79)的抑制潜力。选择具有最佳对接分数的稳定配体-蛋白质复合物,并分析分子动力学轨迹中的关键参数,包括结合稳定性和相互作用,以确定用于治疗开发的MAO-B高潜力抑制剂。结果显示,与天然配体(-11.12 kcal/mol)相比,β-谷甾醇(-11.92 kcal/mol)、角鲨烯(-11.89 kcal/mol)、阿维A酯(-11.46 kcal/mol)、芸香苷(-11.44 kcal/mol)和獐牙菜苷(-11.13 kcal/mol)表现出更高的结合亲和力。另外三种化合物:根皮苷(-11.10 kcal/mol)、土大黄苷(-11.02 kcal/mol)和香叶木素7-O-β-D-吡喃葡萄糖苷(-10.96 kcal/mol)表现出比参考药物更好的结合能力。蛋白质与配体之间的主要相互作用是疏水作用,氢键和π-堆积增强了复合物的稳定性。基于从200 ns MDS得出的几何和热力学指标进行的评估,确定芸香苷、β-谷甾醇和獐牙菜苷为有前景的MAO-B抑制剂。在准生理条件下,这些配体在MAO-B催化位点内的平移和旋转运动最小,表明具有有效抑制作用。保留的热力学可行性强化了这些发现。ADMET分析确定角鲨烯和β-谷甾醇为具有良好药代动力学的中枢神经系统活性候选物,而阿维A酯、芸香苷和獐牙菜苷可能作为外周调节剂,需要进行优化以改善中枢神经系统递送。建议进一步对疗效、药代动力学和安全性进行实验验证,以提高这些命中候选物的治疗潜力。