Compound Synthesis and Management, Discovery Sciences, Biopharmaceuticals R&D, AstraZeneca, 431 83 Mölndal, Sweden.
School of Chemistry, Pharmacy & Pharmacology, University of East Anglia, Norwich Research Park, Norwich NR4 7TJ, UK.
Int J Mol Sci. 2024 Aug 13;25(16):8792. doi: 10.3390/ijms25168792.
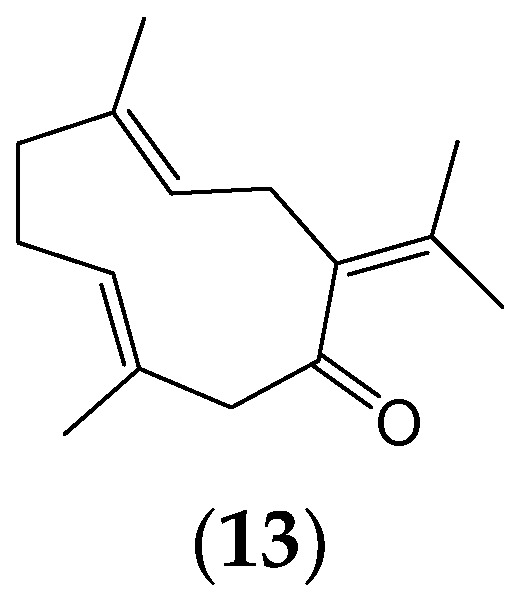
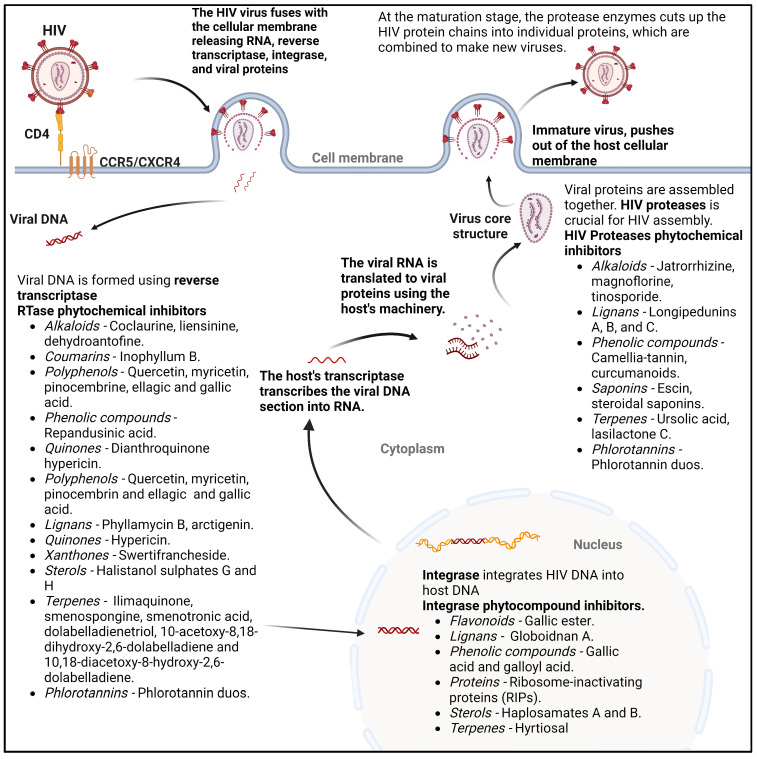
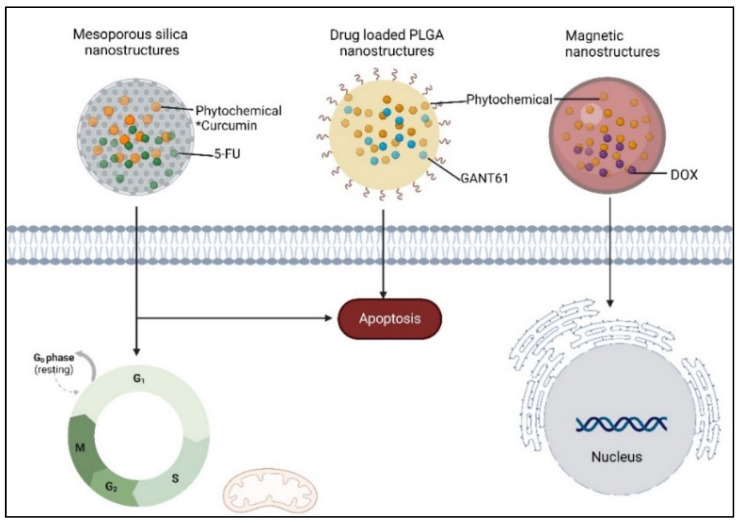
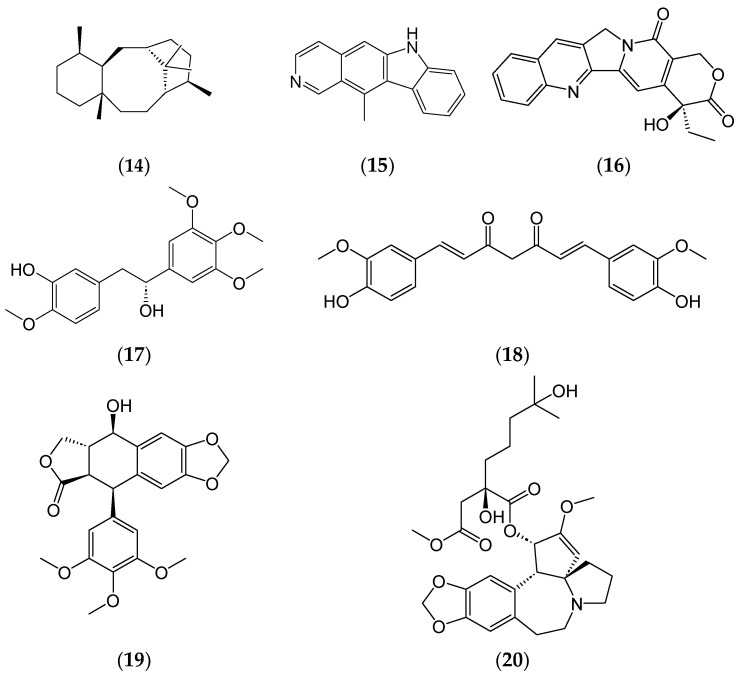
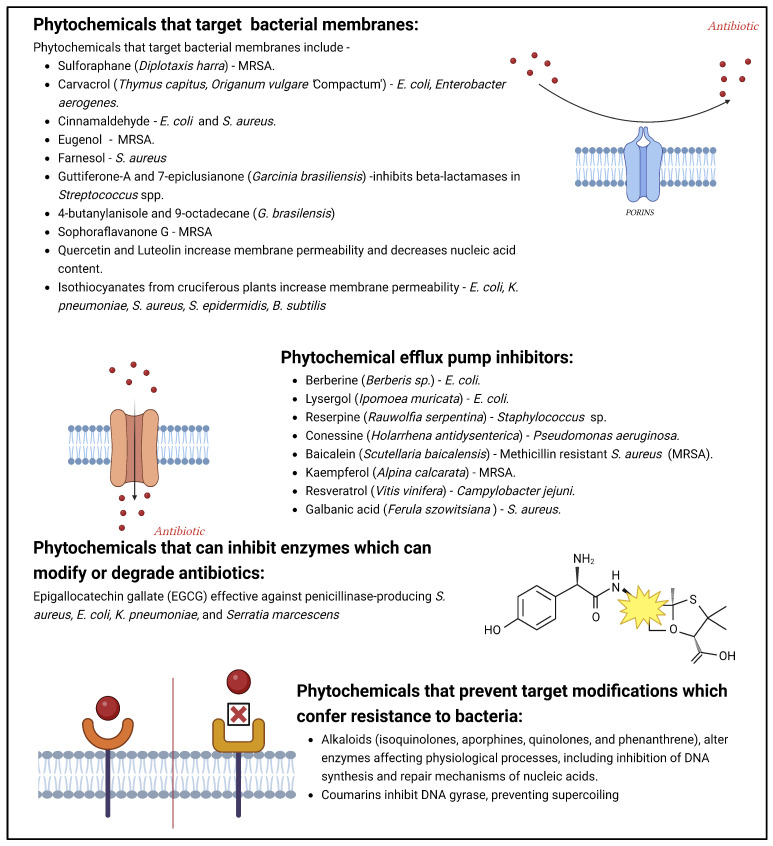
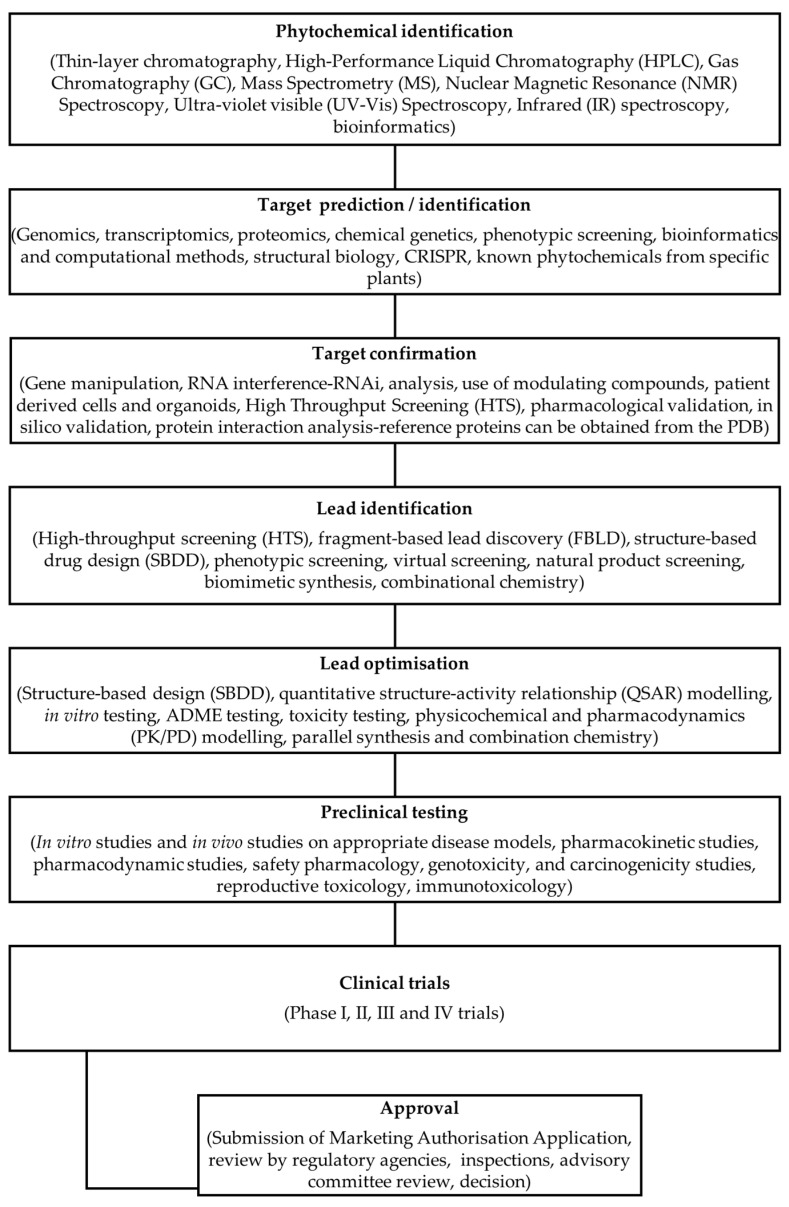
Phytochemicals have a long and successful history in drug discovery. With recent advancements in analytical techniques and methodologies, discovering bioactive leads from natural compounds has become easier. Computational techniques like molecular docking, QSAR modelling and machine learning, and network pharmacology are among the most promising new tools that allow researchers to make predictions concerning natural products' potential targets, thereby guiding experimental validation efforts. Additionally, approaches like LC-MS or LC-NMR speed up compound identification by streamlining analytical processes. Integrating structural and computational biology aids in lead identification, thus providing invaluable information to understand how phytochemicals interact with potential targets in the body. An emerging computational approach is machine learning involving QSAR modelling and deep neural networks that interrelate phytochemical properties with diverse physiological activities such as antimicrobial or anticancer effects.
植物化学物质在药物发现方面有着悠久而成功的历史。随着分析技术和方法的最新进展,从天然化合物中发现生物活性先导物变得更加容易。计算技术,如分子对接、QSAR 建模和机器学习,以及网络药理学,是最有前途的新工具之一,它们允许研究人员对天然产物的潜在靶点进行预测,从而指导实验验证工作。此外,像 LC-MS 或 LC-NMR 这样的方法通过简化分析过程来加速化合物的鉴定。整合结构和计算生物学有助于识别先导化合物,从而为理解植物化学物质如何与体内潜在靶点相互作用提供了宝贵的信息。一种新兴的计算方法是机器学习,涉及 QSAR 建模和深度神经网络,它们将植物化学性质与多种生理活性(如抗菌或抗癌作用)相关联。